Light and the Electromagnetic Spectrum
Light
Light is important in chemistry partly because light can determine structures and properties of molecules. There are many examples of using light for different purposes in our daily lives. For example, microwave oven uses the property of microwave (~100 mm wave length) to boil water. The MRI technique uses radio wave close to analog TV wave (VHF and UHF) in wave length to image the water content in your body. There are many many others.
Electronmagnetic Spectrum
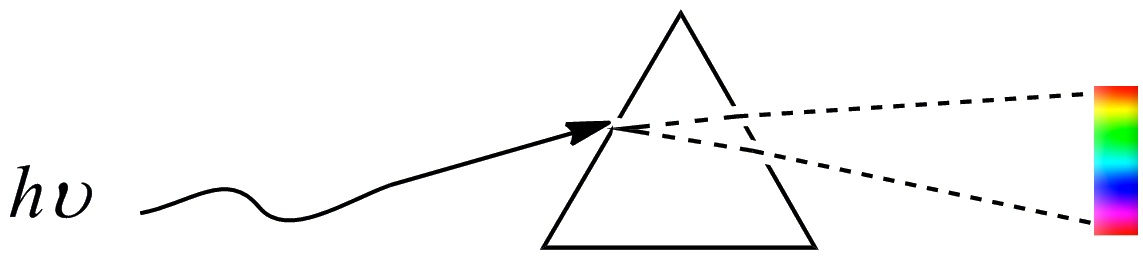
We know what happens when the light passes through a prism. The white light separates into different colors, as shown in the figure below. The separated colors are generally called spectrum.
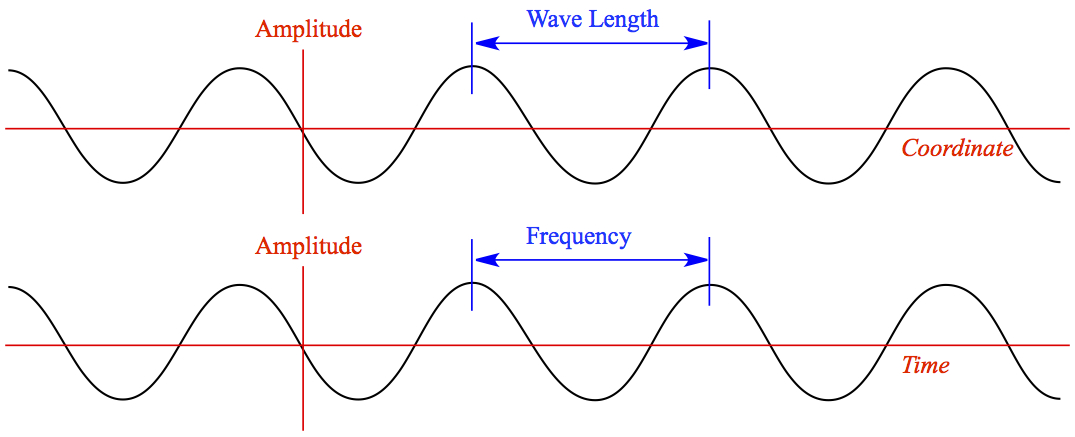
Each color is represented in terms of a wave. There are two characteristics of wave.
- Wave Length (λ) — peak to peak distance (wave in coordinate space)
- Frequency (ν) — # of peaks passing through time(wave in time domain)
The unit of wave length is in the length unit, such as m.
The unit of frequency is in the recipical of time, such as /s (also called Herz or Hz)
Therefore, if these two units are multiplied, it becomes the speed, (length/time). For all electromagnetic waves, the frequency x wave length is always speed of light (2.998 x 108m/s)
It means that λ and ν are in the recipical relationship.
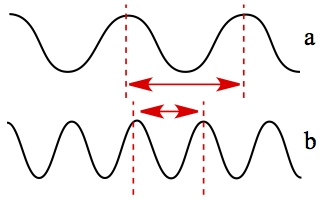
The wave length is longer in case a than that in case b. It also means that case b has higher frequency than in case a.
The following figure shows the length scale of electromagnetic waves with objects of the corresponding size. (Taken from this site.)
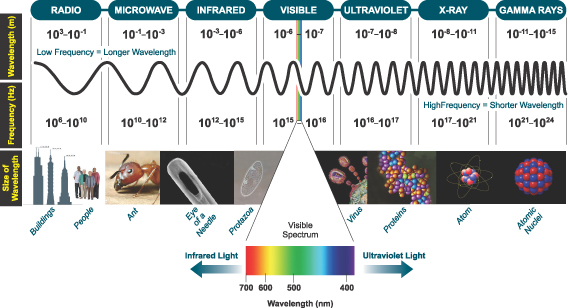
As you can see, the visible light, for which our eyes can detect, is only a very tiny window of the electromagnetic spectrum.
As the name electro-magnetic implies that the wave of this kind is consist of electric field and magnetic field. In a sense, each field induces the other, and pushing each other to propagate the wave. The following figure shows the interweaving of electric field and magnetic field for propagation of light pictorially.
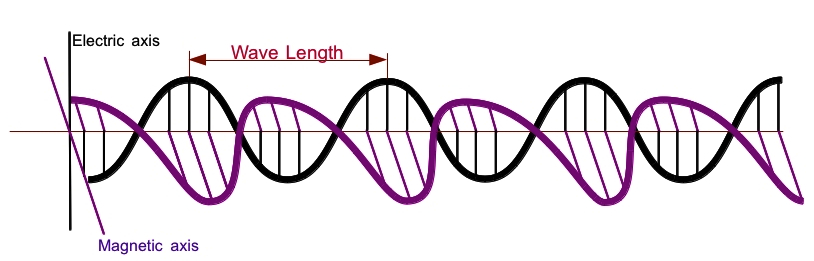
The electric field and magnetic field are perpendicular to each other and the direction of propagation is to the right.
Photoelectric Effect
I just said above that light is a wave. In the following light is explained by a particle. In the photoelectric effect, light is used to eject electron from a metal plate, which travels to the positively charged electrode so that the electrical current is produced. The electric current is registered in an ampmeter.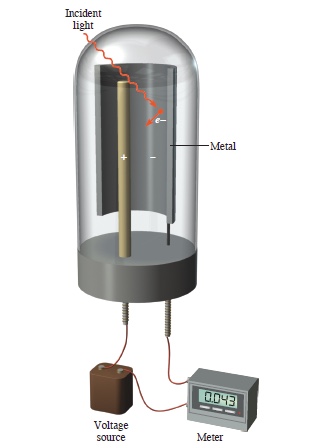
Einstein explained the phenomena by using Planck's idea five years prior, where Planck stated that the energy of a photon, a particle of light, is given by where h is called Planck's constant today and has a value 6.626 x 10-34Js. Using this, Einstein explained the photoelectric effect by where Ek is kinetic energy of the ejected electron and W is so-called work function.
Electromagnetic Energy and Atomic Line Spectra
When the sun's light is passed through a prism, as you know, the color separates. Let's say that you have a precision instrument to separate the color into much more details than a simple prism can do. Upon analyzing the color in the Sun's light, you see the dark band or missing color at certain wave lengths. Such dark lines are called absorption line spectra. The instrument used to observe the spectral lines are something like the following:

Fraunhofer Society
Look at the following figure. This figure is the spectrum of sun's ray taken with the precision instrument in the visible light range.
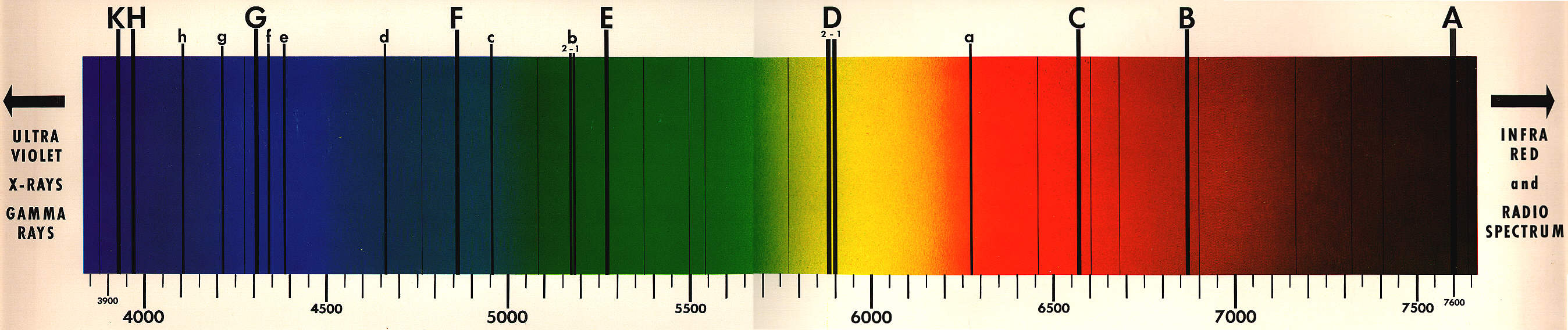
To the right of the figure wave length increases, and the scale is in the unit of Ångström, Å (1 Å = 10-10 m). The dark bands are clearly visible. So, what do the dark lines represent? These lines puzzled the scientists since 1814 (earlier by Wollaston in 1802). However, the mystery was slowly unraveling, starting with the laboratory reproduction of the line spectra.
In the laboratories, experiments similar to the following figure had been done.
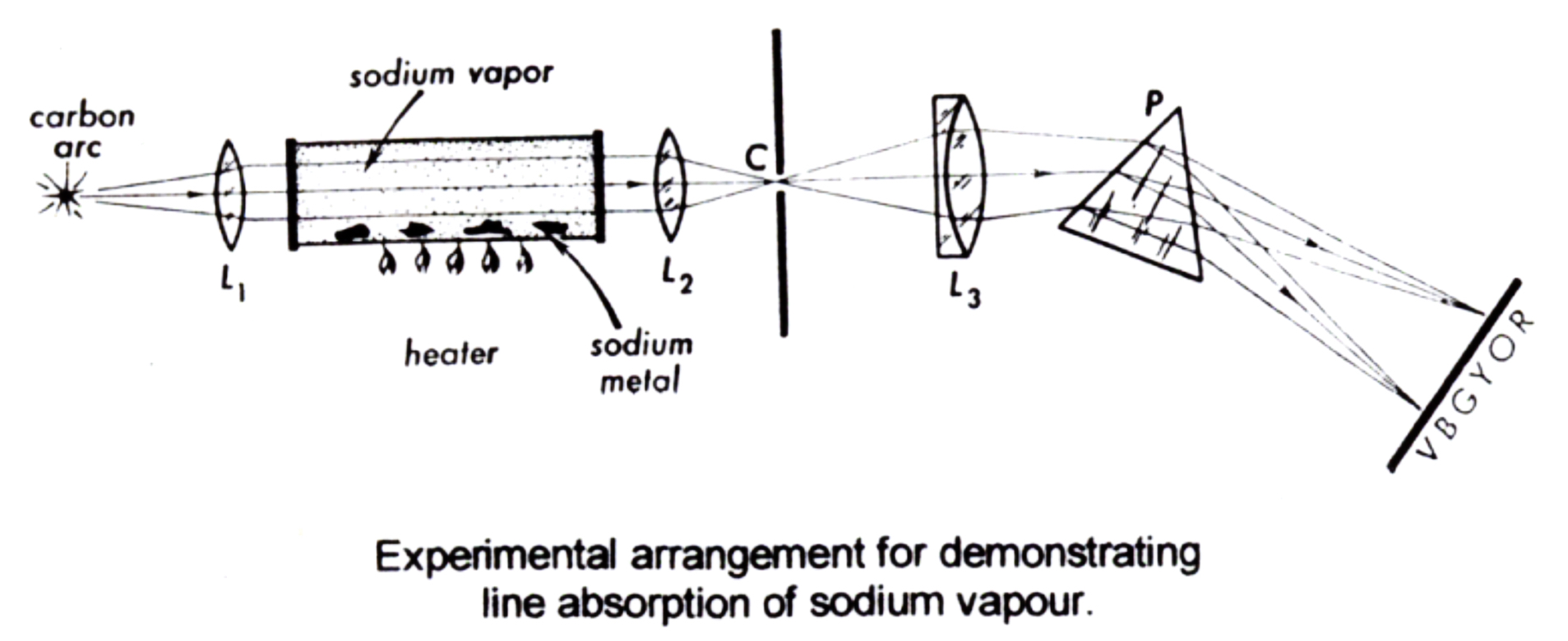
It means that sun is made up, at least partially, of sodium! The consequence of this finding is obvious! The Earth may not be as unique as we thought it was.
Mathematical relationship of wave length with misterious whole number!
Scientists started to find the patterns of other elements in the Sun's ray. What do those pattern tell us? What they started to see, was that there was a relationship between wave length and integers!
Balmer came up with the following, called Balmer equation:
| Wave Length | Frequency |
|---|---|
In these equations R∞ is what is known today as Rydberg constant (1.097 x 10-2 nm-1), c is the speed of light, and n is an integer larger than 2. Be careful with conversion, as R∞ unit is different from c! In the frequency equation, the extra factor of 109 nm/m is used to convert the nm-1 unit in the R∞ into m-1 unit so that the frequency comes out in the Hz (s-1) unit.
Rydberg then generalized Balmer's equation to fit all line spectra of hydrogen. Thus, we have the Balmer-Rydberg equation
| Wave Length | Frequency |
|---|---|
with m < n.
It works great with hydrogen atom, but nothing else. They have to wait another 30 years or so to fully comprehend where the atomic spectral lines come from. However, some partial answers had started to come out, particularly the relationship between the line spectra and the structures of atom by Niels Bohr.
Particle-like Properties of Electromagnetic Energy
So, we said above that electromagnetic wave is a wave with frequency and wave length. It is a wave! Einstein, however, said in 1905 (annus mirabilis) that he could explain the phenomenon called "photoelectric effect" if he used light being particles! Einstein used the idea from Planck whose seemingly desparate theory to explain the so-called black-body radiation through a packet of energy (particle of energy), called a quantum.
In his theory, Planck considered the energy of a light particle, called photon, depends on the frequency of light, given by the following equation.
where h is Planck constant (h = 6.626 x 10-34Js, very tiny number!), and since c = λ ν, the energy of light particle is associated with the wave length, as
What's important here is that the electromagnetic radiation is both wave and particle, wave-particle duality.
Tentative structure of atom: Bohr atom
Physicists then wanted to understand why atom is stable. At the time,
they only new atom has a heavy nucleus with + charge and electrons with -
charge, are around nucleus, according to
Rutherford (Chapter 2). You know that
+ charge and - charge attract each other. If an electron with a negative
charge is zipping initially around
a nucleus with positive charge, the electron should
start to spiral down to nucleus by emitting light of higher and higher
frequency, as speeding up toward the nucleus. And, eventually, the electron
would collide with nucleus, and the atom would disappear.
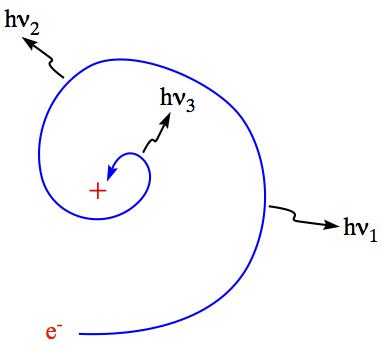 This is the scenerio that the classical electromagnetism tells us.
(See the figure on right.)
This is the scenerio that the classical electromagnetism tells us.
(See the figure on right.)
What it means is that atom is not stable in terms of classical physics! It also means that we needed a new and fundamental mechanics for atomic and subatomic processes!
Bohr in 1914 gave the partial answer to the structure of atom, and simultaneously solved hydrogen atomic spectral lines. His idea was hinted from Rutherford's nuclear atom. Bohr was able dervie an mathematical expression for state of hydrogen atom, as having a number of definite and stable orbits that are permanent. Electrons in atoms then circumnavigate the nucleus on these orbits. The state of hydrogen atom is decided by a single integer, n, as shown below,
where RH is called Rydberg constant (1312 kJ/mol, or 2.18 x 10-18J).
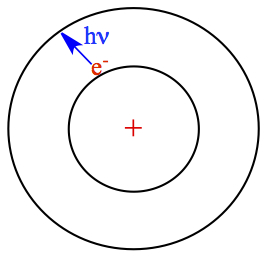
The figure on left shows the atomic structure according to Bohr.
Since there are
stable orbits, and in order for the electron to go to higher orbit, say
n = 2 state (or
any other n except for n=1 is called excited state
), from its
lowest energy state (ground state with n = 1),
the jump (it is called transition) must be
accompanied by an absorption of
a photon of energy with E = h ν. This jump is what is known as
quantum jump! Atom absorbs specific frequency of light to make a quantum jump.
Now, we can explain why Balmer-Rydberg equation only has integers. According to Bohr, the difference between the energies of the two states correspond to the quantum jump with photon energy, Ephoton = h ν = ΔEnm. The last term, ΔEnm is the difference between the two states where m labels the lower energy of the two states, and n labels the higher.
This equation completely solves the atomic spectra of hydrogen.
Example: What is the energy in kJ/mol of photon being absorbed for transition between n = 1 and n = 2?
Using the above equation, we have
we have m = 1 and n = 2. Since the answer is expressed by per mole quantity, we need to multiply the above by Avogadro's number.
Place all the constants in with units, we have a
Depending on the lower states of the two integers in the Balmer-Rydberg equation, the state with label m, the spectra are assigned as spectral series.
| Name | Lower state | Region |
|---|---|---|
| Lyman Series | m = 1 | Ultraviolet |
| Balmer Series | m = 2 | Visible |
| Paschen Series | m = 3 | Infrared |
| Brackett Series | m = 4 | Infrared |
| Pfund Series | m = 5 | Infrared |
| Humphreys Series | m = 6 | Infrared |
In the Balmer series, the lower state, m = 2. Since we have the energy of photon absorbed, all we need to do is to rearrange the following equation and solve for n,
for which we know Ephoton, and RH. To isolate n, you need to do the following, Here we know m = 2, then, Then, the recipical of the equation is, Therefore, the transition was from m = 2 to the n = 5 state.
Wave-like Properties of Matter
Having seen wave is described by particle, de Broglie thought that the opposite might be true. It means that, this tiime, particle can be considered as wave. Oh, why not!? So, he derived an equation relating the characteristics of wave and particle in the following way:
We already established that
Therefore,
and from Einstein's E = mc2, we have
Since c, the speed of light, is a velocity, we can substitute v, velocity of particle, in place of c. The denominator in the last term, mv, is a linear momentum, which is associated with particle of mass m moving at the speed v. Weird cool, right?
The speed of light is 2.998 x 108 m/s, and the mass of an electron is 9.11 x 10-31 kg.
The symbol Å, angstrom, is a distance unit of 10-10 m. So, this wave length corresponds to the atomic scale. Since the average radius of hydrogen atom is 0.529 Å, the circumnavigation of electron is 3.32 Å and matches with our calculated value.
As for Usain Bolt, the calculation is straight forward. His wave length is then,
So, for a macroscopic particle, the de Broglie wave length is very very small. The effect of wave nature of us is not even detectable; it is even smaller than the atomic scale!! On the other hand, for a hydrogen atom, the wave length is the same scale as the atomic scale, and therefore the duality of wave and particle exists.
Quantum Mechanics and the Heisenberg Uncertainty Principle
So far, we have established:
- All electromagnetic waves for different purposes (such as nuking your
coffee or getting chest X-ray), differ only by wave
length.
- Wave is particle.
- Particle is wave.
Heisenberg's Uncertainty Principle
Weirdness doesn't stop here. One of the corner stones of quantum mechanics is Uncertainty principle of Heisenberg. The principle sharply separates the ideas of the classical world (pre-quantum era) and the quantum world.
When a baseball is thrown out of the hand of a pitcher, you can track how the ball is traveling. If you are a good baseball player and if you know what kind of pitch the pitcher would throw, you can perhaps hit the ball hard. This is partly because you can track the ball with your eyes and your brain computes the ball's trajectory. The trajectory is a notion completely mathematically definable using initial speed and direction (velocity), as well as initial position of the ball. So, trajectory needs position and velocity. This is classical world.
In a microscopic world the notion of trajectory must be rejected, said Heisenberg. Here he establishes one of the most fundamental aspect of the quantum world through his Uncertainty Principle.
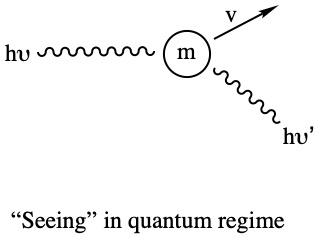
Heisenberg's idea is that the motion of quantum particles, such as electrons, protons, photons, etc., are fuzzy. If you know more about the position, you would lose the exactness of speed and its direction. On the other hand, if you know more about the velocity (speed and direction), you know less about where the particle is. That's way too weird!! Anyway, Heisenberg put his idea into mathematical form:
It tells you that the products of uncertainty in measurement of coordinate, Δx, and uncertainty in the measurement of momentum (velocity), Δmv is always greater or equal to Planck's constant, h/4π. This inequality gives the fuzziness in the motion of subatomic particles.
Weird!
Wave Functions and Quantum Numbers
Mathematical foundation of quantum mechanics was layed by Heisenberg in 1924. Soon after, Heisenberg's advisor and his colleague quickly established what is known today as matrix mechanics. In 1926, Schrödinger established his so-called wave mechanics. It was argued among the physcists of the day which one is correct formulation of quantum mechanics. Schrödinger later showed the equivalency of the two formulations. Here is his equation that bares his name,| Schrödinger Equation |
Quantum mechanics can be applied to different systems at substomic and molecular levels. Since in this chapter we are dealing with atomic structure, we are going to look at the Schrödinger's result to hydrogen atom and its spectra.
When solving the Schrödinger equation, one obtains 3 sets of integers and a set of half-integers, called quantum numbers.
- 1. Principal Quantum Number, n
- 2. Angular Momentum Quantum Number, l
- 3. Magnetic Quantum Number, ml
- 4. Spin Quantum Number, ms
What's interesting about these sets of numbers, is that they describe the structure of atoms. These numbers completely describe the state of atom.
Principal Quantum Number, n
This quantum number describes the energy of the hydrogen atom. This formulation is completely different from that of Bohr, seen above, yet the constant RH turned out to be exactly the same as the one obtained by Bohr. As before, the value for RH = 1312 kJ/mol for hydrogen atom.
Angular Momentum Quantum Number, l
Shape of orbital is determined by anuglar momentum quantum number, l. Orbital is related to the probability of finding an electron. If one squares the orbital, it becomes probability.
The value of l runs from 0 to n - 1. So, if you start with n = 2, l can run from 0 to 1. If n = 5, then l can be 0, 1, 2, 3, or 4.
- If l = 0, the orbital is said to be s orbital.
- If l = 1, the orbital is said to be p orbital.
- If l = 2, the orbital is said to be d orbital.
- If l = 3, the orbital is said to be f orbital.
- If l = 4, the orbital is said to be g orbital.
These different orbitals have different shapes. We'll look at them in the next section.
Magnetic Quantum Number, ml
Degeneracies in the angular momentum quantum numbers —meaning that how many of the same orbital in the given l quantum number.
There are (l - 1)*2 sets of orbitals in a given l, and the value of ml runs from -l to +l in itegral manner.
So,
- if l = 0, ml = 0
- if l = 1, ml = -1, 0, or +1
- if l = 2, ml = -2, -1, 0, 1, or 2
- if l = 3, ml = -3, -2, -1, 0, 1, 2 or 3
- if l = 4, ml = -4, -3, -2, -1, 0, 1, 2, 3,
or 4
This quantum number came about because the atomic spectra split when the atom is in magnetic field. This splitting occurs due to the negatively charged electron moving around the nucleus. The magnetic field is created when there is a moving charge. (Look at induction of magnetic field by current in the wire, here. Video is not a very good quality, but very informative.)
Spin Quantum Number, ms
This spin quantum number only assumes either +½ or -½. The +½ electron is called α (alpha) electron, and the -½ electron is called β (beta) electron, represented as ↿ (half up arrow) and ⇂ (half down arrow), respectively, when used within the orbital energy diagram, as discussed below.
Notations
Using above mentioned quantum numbers, we can find out all possible combination of n, l, ml and are listed in Table 5.1 below.
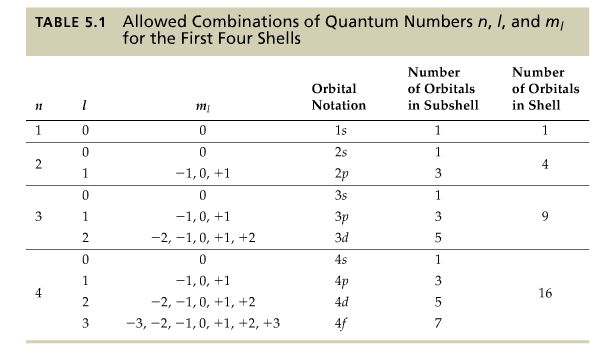
If n = 1, only s orbital is possible, and the orbital arising from the n and l, we call it 1s orbital, etc, as shown in the table.
As it is seen, all p orbitals have 3 sets of orbitals, all d orbtials 5 sets, all f orbitals 7 sets.
Energies of orbitals are shown in the following figure.
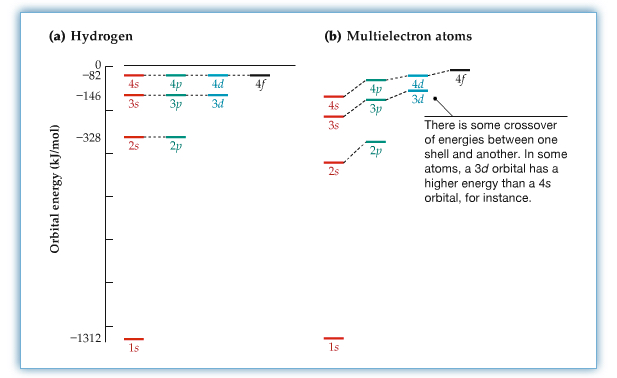
For hydrogen atom the energy of the orbital within the same principal quantum numbers are the same, while multi-electron case are different, due to electron-electron interactions in the atom.
Shapes of Orbitals
What orital represents is where the electrons are found. Each orbital can hold either 0, 1 or 2 electrons. So, for any s orbital can hold up to two electrons, but any p orbitals can hold up to 6 electrons since there are three sets of p orbitals due to the values of ml (-1, 0, and +1). Therefore, d orbitals → 10 electrons maximum since ml = -2, -1, 0, 1, 2.
Shapes of orbitals are depended upon the value of l quantum number.
s orbitals for l = 0 — spherical
p orbitals for l = 1
d orbitals for l = 2
f orbitals for l = 3
The following beautiful pictures of orbitals show the shapes of each and the shell structure is clearly shown. The picture is laid out according to the Periodic Table of Elements.

Electron Spin and the Pauli Exclusion Principle
As mentioned above, there are half-integral quantum number, called spin quantum number, ms. This quantum number also describe about magnetic field of electron. However, this has nothing to do with motion of electron around nucleus. Electron seems to behave as ultimately tiny magnet. The two quantum numbers, ±½, can be thought of as N---S and S---N configurations of a magnet.
In 1925, after very careful sorting of elements of on the Periodic Table, Pauli realized that all 4 sets of quantum numbers, n, l, ml, and ms must be different for each of the electrons in an atom. This is called Pauli exclusion principle.
Another way to put this principle, is that there are no more than 2 electron can be placed in an orbital. Therefore, 1s orbital can have maximum of 2 electrons, 2p—6 electrons, 3d—10 electrons.
Orbital Energies in Multielectron Atoms
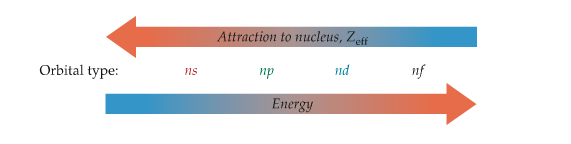
As shown in the Wave Function and Quantum Number section , the orbital energies of multielectron atoms are different from that of the hydrogen atom. This is due to the fact that electron in the low-energy orbitals shields outer electrons. If outer electron is shielded, the attraction to nucleus from the outer electron is effectively less (you can call it effective nuclear charge). The effectiveness of shielding depends on the types of orbitals according to the following figure.
Electron Configurations of Multielectron Atoms
Atoms in ambient temperature and pressure are assumed to be in its lowest energy state, called ground state. When external source such as light or heat is given to atoms, atoms may go to what is known as excited state. Excited state has finite life time, and eventually go down to its ground state. Here we look at how the ground state of atoms is like by placinig electrons into orbitals.
Electron Configuration—how electrons are placed in atomic orbitals. For example, 6-electron atom, carbon is written as
Use Aufbau Principle. Given the principal quantum number, you can write all possible angular momentum terms, as follows. If n = 1, then orbital arising from it is 1s. If n = 2, then orbitals arising from it are 1s, 2s, and 2p. If n = 3, then orbitals arising from it are 1s, 2s, 2p, 3s, 3p, and 3d. And so on so forth.
Then cross out anti-diagonally, as follows.
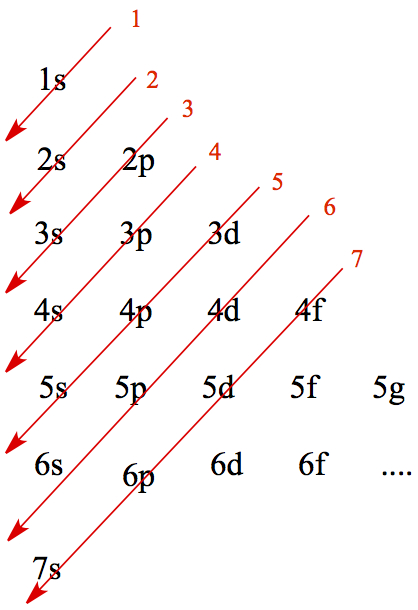
Following the red arrow in order, and write down the orbitals. This is the order to which you fill electrons in multi-electron atoms.
Then, ordering of orbitals are:
As you can see for carbon, the electron configuration is s22s22p2. The superscripted numbers tell how many electrons there are in the orbitals.
Here are some rules you must follow to construct electron configuration:
- Lower energy orbitals are filled first.
- An orbital can hold two electrons of opposite spin (ms=
+½ and -½)
- For degenerate orbitals (such as p, d and f orbitals),
use Hund's rule.
- One electron goes into each orbital until all are half-filled.
| Z | Name | e- config | Orbital Energy Diagram |
|---|---|---|---|
| 1 | H | 1s1 | |
| 2 | He | 1s2 | |
| 3 | Li | 1s22s1 | 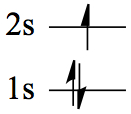 |
| 4 | Be | 1s22s2 | 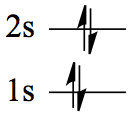 |
| 5 | B | 1s22s2 2p1 | 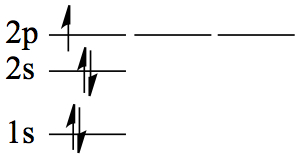 |
| 6 | C | 1s22s2 2p2 | 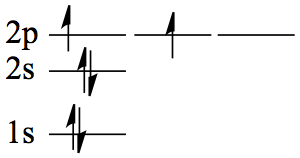 |
| 7 | N | 1s22s2 2p3 | 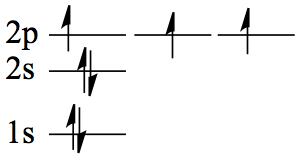 |
| 8 | O | 1s22s2 2p4 | 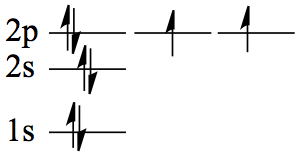 |
| 9 | F | 1s22s2 2p5 | 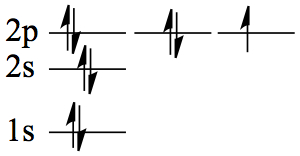 |
| 10 | Ne | 1s22s2 2p6 | 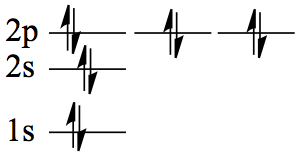 |
| 11 | Na | 1s22s2 2p63s1 | 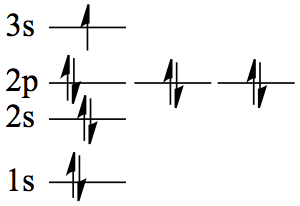 |
Do you see the pattern? When constructing orbital diagram, don't forget the Hund's rule!
Anomalous Electron Configurations
Even though, the Aufbau principle works great, there are some exceptions that you have to watch out. For example, Cr (z=24) and Cu (z=29). The expected electron configurations of these atoms are:
For Cu, 1s22s2 2p63s2 3p64s2 3d9, but observed one is 1s22s2 2p63s2 3p64s1 3d10
So, as you can see in both cases, s orbital is made into singly occupied, and d into either half-filled or completely filled. The half-filled configuration is more stable than s filled and d unevenly filled.
Here, all anomalous electron configuration species are listed:
Cr, Cu, Mo, Au, Ru, Rh Rg, Pt, Pd, La, Ce, Gd, Ac, Th, Pa, U, Np, Cm, Ds, and Rg.
Electron Configurations and the Periodic Table
Now, we are at the point for which how the periodic table is layed out. It might be quite obvious now. Look at the following figure.
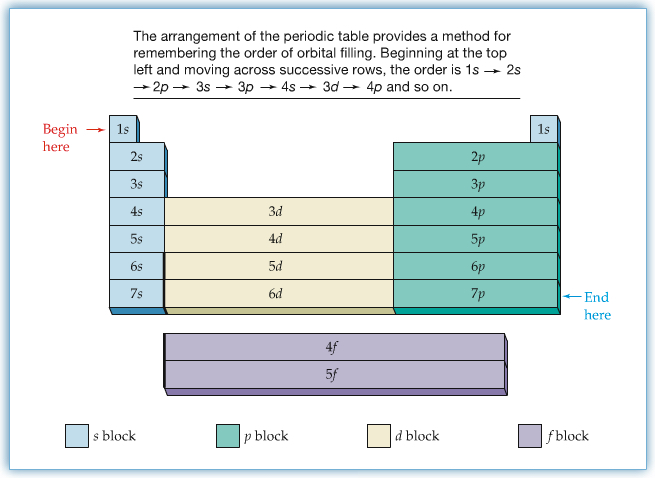
From quantum mechanical point of view, each row represents the principal quantum numbers, and if the element belong to the same group, the highest energy orbitals have the same characters.
| Group | Orbitals |
|---|---|
| 1 and 2 | s |
| 13 to 18 (or 3A to 8A) | p |
| 3 to 12 (transition metals) | d |
| Lanthanides and Actinides | f |
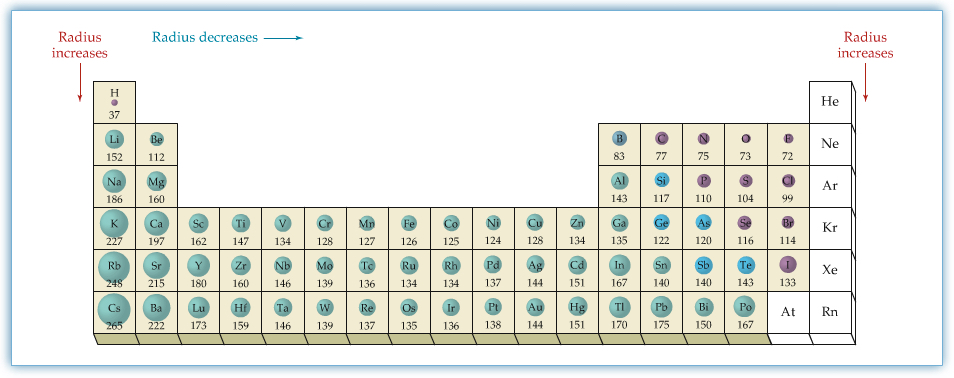
Atomic Radii
Atomic Radius—defined by the half of the distance between two of the same nuclei (homonuclear diatomic molecule).
Hydrogen molecule, H2, has its bond distance of 0.741 Å. Therefore, hydrogen's atomic radius is 0.375 Å. The following table shows the atomic radii. The values are in pm.
Electron Configurations of Ions
As we have seen in the previous chapter, electron configuration is the key in understanding where atoms are located on the Periodic Table of Elements.
Can we understand how ions are made in terms of electron configuration?
We know that Na atom tends to become +1 ion, and the group 17 atoms tend to become -1 ions.
| Group | Charge |
|---|---|
| 1 | +1 |
| 2 | +2 |
| 17 | -1 |
| 16 | -2 |
| 15 | -3 |
The electron configurations of Na (11 electrons) and Na+ (10 electrons) according to aufbau principle are:
Na: 1s22s22p63s1
Na+: 1s22s22p6
The electron configurations of Cl (17 electrons) and Cl- (18 electrons) according to aufbau principle are:
Cl: 1s22s22p63s23p5
Cl-: 1s22s22p63s23p6
The point here is that ions are made such that the noble gas configuration is obtained for each. For Na, Na+ assumes Ne configuration, and for Cl, Cl- assumes Ar configuration.
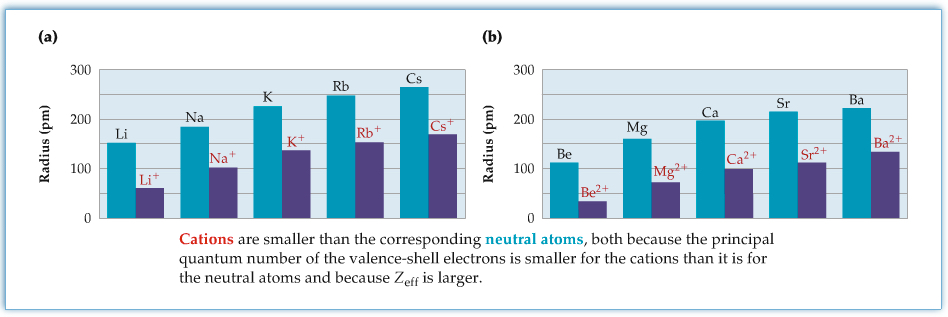
Ionic Radii
Ionic radii of atoms are substantially different from those of neutral atoms. The following two figures show, respectively, ionic radii of cations (groups 1 and 2), and ioinic radii of anions of halides (group 17). The cationic radii are much smaller than the parent neutral atoms, and the ionic radii of anions are much larger than the parent. The effect has to do with the effective nuclear charge that we talked about in Chapter 5. For cation, the valence electrons are less shielded due to the loss of electron such that there are more positive charge effectively. On the other hand, the anion's valence electrons are more shielded due to extra electron.

Ionization Energy
Ionization energy (or ionization potential)—energy required to remove the highest energy electron from atom to make a cation. We denote it as Ei1.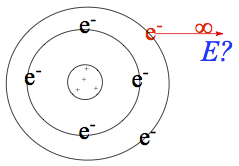
We know from the structure of hydrogen atom that its ionization energy is 1312 kJ/mol (Chapter 5).
Second ionization energy is the energy required to remove the second electron from a cation, denoted as Ei2.
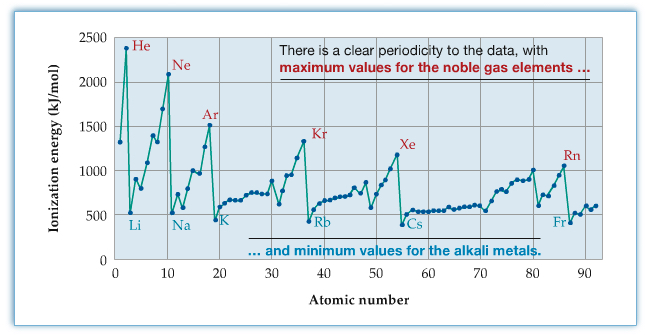
The periodic trend of ionization enery is shown in the following figure.
Higher-order ionization energy, that is the energy required to remove 2nd, 3rd and higher electrons, also has some trend. The following table shows the trend.
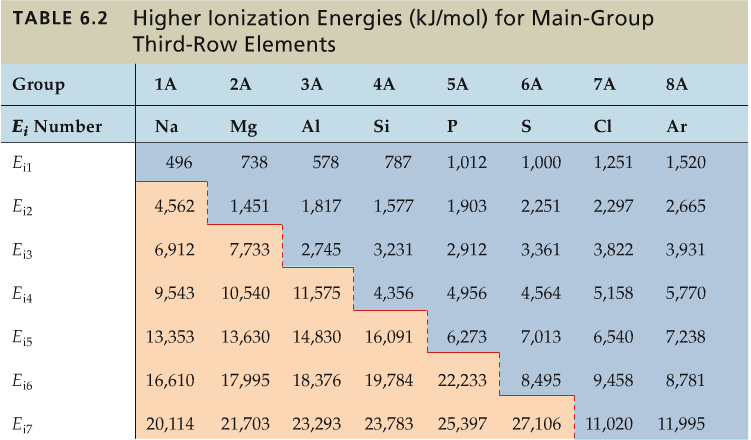
There is a shape dividing line between relatively small ionization energy and very large ones. For Na, after Ei1, the ionization energy sharply rises. For Mg, it is after two electrons. For Al, 3, and so on so forth.
Electron Affinity
Electron affinity—energy released by atom upon receiving an electron.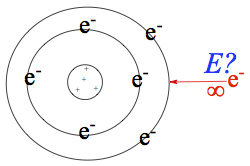
If the atom such as Cl receives an electron and becomes Cl-, the resulting anion is more stable by more than 350 kJ/mol! If unstable anion is formed, then we define electron affinity to be 0 kJ/mol. This is due to the fact that experimentally such energy is not measurable.
The following figure shows the periodic trend for electron affinity.
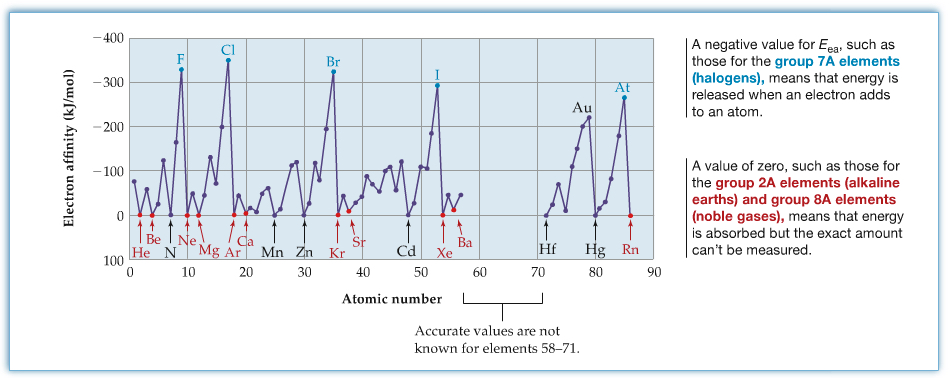
The periodic trend is that, for main group elments, the electron affinity increases as the atomic number, Z, increases. However, as you can see in the figure, nitrogen and phosphorus have the decreased of electron affinity. This is due to the stability of these atoms for their electron configurations being half-filled with the p3 configuration. You see the similar trend in Mn where the electron configuration is ...4s23d5.
Octet Rule
From what we have seen on the periodic trend is that:
- Group 1 elements have the low 1st ionization energy
- Group 2 elemnets have the relatively low value up to Ei2
- Group 17 elements have the large electron affinity — therefore more
stable
- Group 18 elements have the largest ionization energies, and zero
electron affinity — both tell stability is hight in these elements
All of the above facts shows the stability of noble gas electron configurations in the main group elemetns.
Octet Rule:
So, whenever possible, the main group atoms tend to surround themselves
with eight electrons to become the noble gas electron configuration.
All the ions have 18 electrons, therefore, they are like Ar. For Ca, Sc, and Ti ions, the ionic charge increases, and it means that electrons feel more attraction to the positive charges. Therefore, among the three, from small to large, you have Ti4+ < Sc3+ < Ca2+.
The atomic radius of S is listed as 104pm in the figure above for the atomic radii. Comparing this number with the ionic radius of the cations shown above Ca2+ ion is about 100pm. Therefore, the negative ion of S should be a lot larger than its neutral atom. Thus, the ordering of size must then be:
Lattice Energy
Lattice energy is the stability (in + value) of formation of ionic compound from the constituent ions in gas phase. For example, in the case of the LiF(s), the lattice energy is given by chemical equation,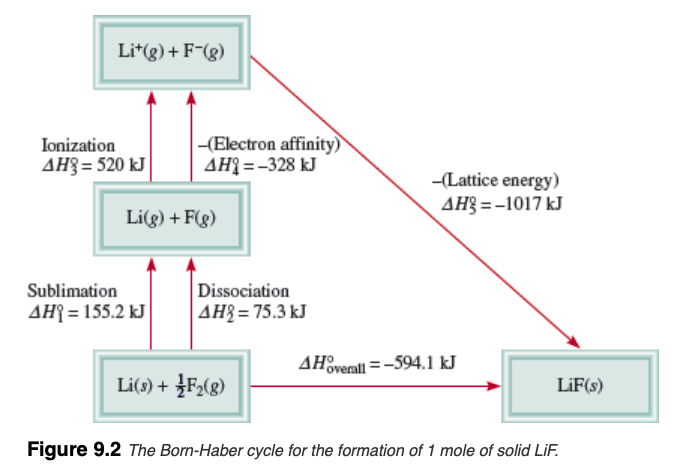
Covalent Bond
When molecules are made, chemical bonds formed between different nuclei. The chemical bonds formed are so-called, covalent bonds. A covalent bond is formed between two nuclei so that resulting molecule is stabilizized, hence existence of the molecule.The interactions of particles in H2 molecule for example is only electromagnetic in nature, more specifically Coulomb interactions. As shown below, even a simple molecule such as H2 has this many interactions to be taken into account, although they are all Coulombic in nature,
where V is the interaction energy between two charged particles, ei and ej, rij is the distance between the charged particles i and j, and c is the proportionality constant. If the two charges are the same V > 0, which means that the interaction is repulsive. On the other hand, the two charges are different, V < 0, which means that the interaction is attractive. In H2, we have these interactions:
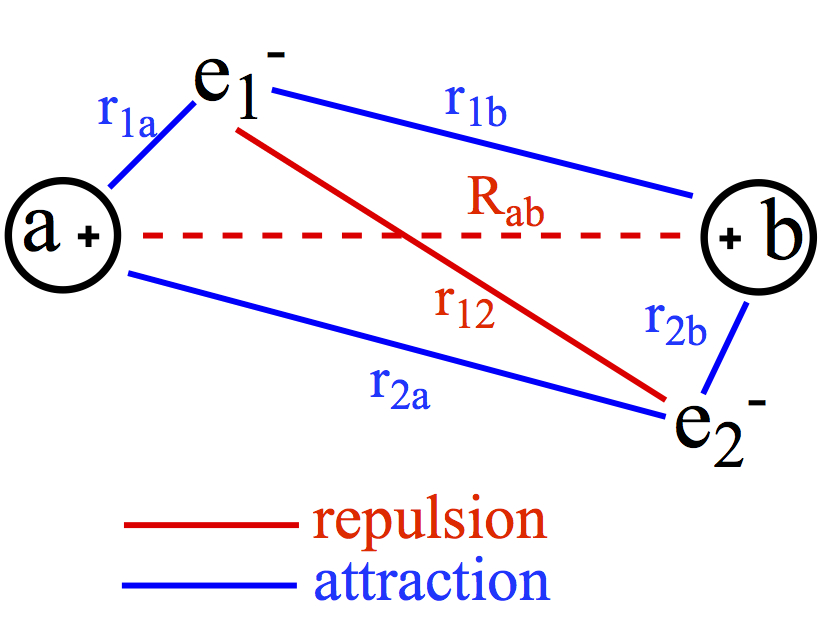
The red lines are repulsive interactions, and the blue lines represent attractive interaction. As simple as it seems, in H2 molecule has six interactions to worry about. When we put these interactions in Schrödinger equation, we obtain the quantum mechanical description of chemical bond.
Remember from the previous chapters that Schrödinger's theory is wave mechanics. So in order for two H atoms to form a chemical bond, two waves one from each H atom collide. The figure below shows what happens when the two hydrogen atoms collide. Notice the tails of the two wave functions overlapping.
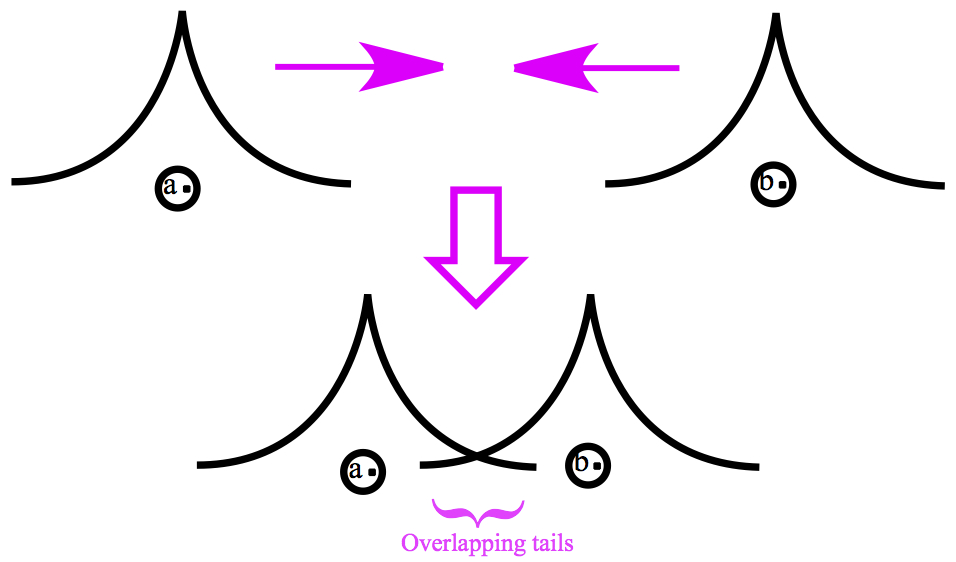
Afterall, these are waves. When, for example, two waves on water collide, the resulting wave shows interference pattern. Similarly, interference does occur in quantum mechanics, as well. Two types of interference: 1) constructive and 2)destructive. Constructive interference in H2 makes a bonding orbital, and destructive interference results in the so-called anti-bonding orbitals. (More on this later.) Basically, the bonding characteristic of covalent bond came from the interferences of two colliding wave functions.
As far as energy is concerned, when chemical bond forms, the molecule is stabilized. The stability of molecule, as compared to two separated atoms, tells that the energy is released when chemical bond forms. On the other hand, when you want a chemical bond to break, you need to inject energy into the bond. The relationship between energy of molecule and the bond distance between two nuclei is shown in the figure below.
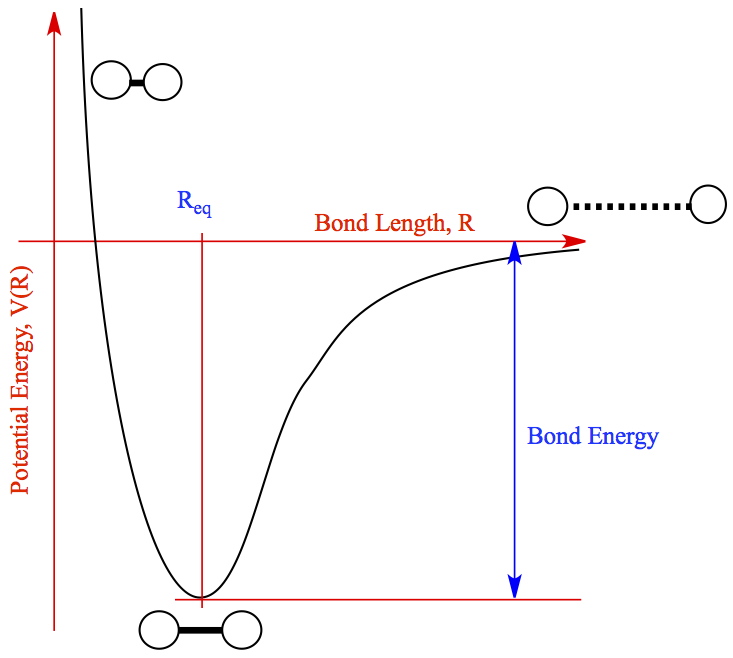
The general shape of the energy of any molecule as a function of bond distance looks like the above; different bonds have the different bond energy and the location of the equilibrium bond distance, Req, shown at the bottom of the well in the figure. The equilibrim bond distance is what we generally refer to as bond length or "bond distance".
So, from Req and try to break a covalent bond, you need to add energy to the system, while from two separated atoms to form a bond, there would be an energy release.
Strength of Covalent Bonds
It is possible for a molecule to have more than one covalent bond between the same nuclei. We generally refer to them as multiple bonds. If there are two, it is said that there is a double bond, triple bond for three, and so forth. For example, nitrogen molecule, N2, has a triple bond.

So, as shown in the figure above (energy of molecule as a fxn of bond distance), one needs to put energy into the molecule to break a chemical bond. If there is more than one bond between the two nuclei, it stands to reason that there are more energy needed to break the multiple bond. Here, some examples are presented.
| # of bonds | CC | NN | CO |
|---|---|---|---|
| Single | 350 | 240 | 350 |
| Double | 611 | 418 | 732 |
| Triple | 835 | 945 |
Polar Covalent Bonds: Electronegativity
When a chemical bond is formed, the electrons feel their environment. If the bond formation dealt between Na and Cl, due to Na's low ionization energy and Cl's high electron affinity, Cl grabs an electron from Na's valence shell to form Cl- ion and Na becomes Na+. In this case, the bonding is ionic. In the case of two carbon atoms, the resulting chemical bond is covalent in nature, and since both nuclei are C, the two C atoms share the two electron equally. However, there are many many molecules that their situations are in between NaCl and -CC- bondings. We refer to covalent bonding between the two extremes as polar covalent bond. A figure below shows the situation.

In the figure above, δ indicate a partial charge of atom in the molecule. The δ+ indicates that the atom has partiall positive charge, and the δ- indicates that the atom has partial negative charge. Even in a NaCl molecule, the overall charge of the molecule is zero, however, because of high electron affinity of Cl and the low ionization energy of Na, Cl gets - nad Na gets +. Similarly, in molecule in general has partial charges on atoms involved in bonding. One way to understand the polarity in covalent bonds, electronegativity can be used to assign partial charges on atoms. There are periodic trends in elelctronegativity, as shown in the figure below.
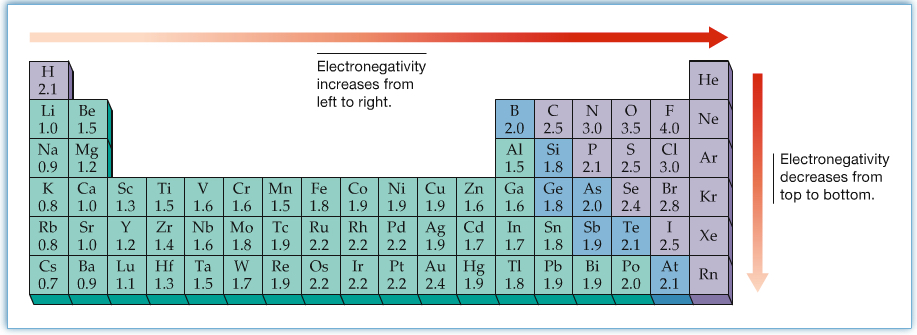
One can define electronegativity to be the ability of atom in a molecule to attract the shared electrons in a covalent bond. The highest electronegativity is assigned for fluorine, and the value of electronegativity decreases as going down the row in the periodic table, as well as going toward the left-side of the table.
-
If the electronegativity difference, ΔEN, between the two atoms
involved in the bonding is
- ΔEN > 2, then the bond is ionic
- ΔEN < 2, then the bond is polar covalent
One of the consequences of having a polar bond in a molecule is that, among other things, it can predict reactivity of molecules, particularly in organic chemistry that you take next year!
Electron-Dot Structures, Lewis Dot Structures
Covalent bond is to share two electrons between two nuclei. So, one can draw H2 molecule as the following.
The two dots in the middle indicate that two H's share two electrons. This is called electron-dot structure or more generally known as Lewis (dot) structure.
In order for us to draw a Lewis structure for a given formula, one must know the number of valence electrons. For the main group elements and hydrogen, we have:
| Group | # valence e- | Example | # of bonds |
|---|---|---|---|
| 13 | 3 | 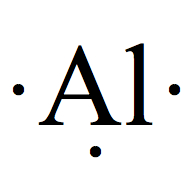 | 3 |
| 14 | 4 | 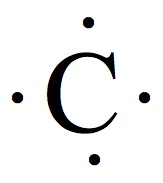 | 4 |
| 15 | 5 |  | 3 |
| 16 | 6 | 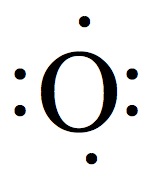 | 2 |
| 17 | 7 |  | 1 |
| H | 1 | 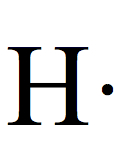 | 1 |
We now utilize the octet rule we learned in Chapter 6 to draw a Lewis structure. Let's use water as an example. For H2O, if we do the following all electrons are paired, and more importantly, oxgen atom satisfies octet.
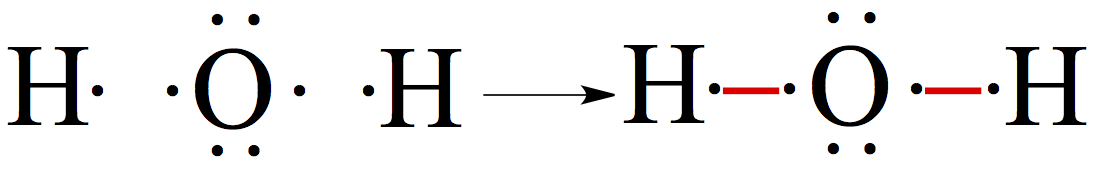
If we place the oxygen atom in the middle and two hydrogens on the side, you can connect two dots that are not paired (red lines). The line represents the covalent bond. Then, oxygen now satisfies octet, having 8 electrons around it.
You notice that the resulting water structure has two pairs electrons, these pairs of electrons are called lone pair. The lone pair electrons in general don't usually react to form a bond to another atom.
So, drawing a Lewis structure basically boils down to connecting two dots.
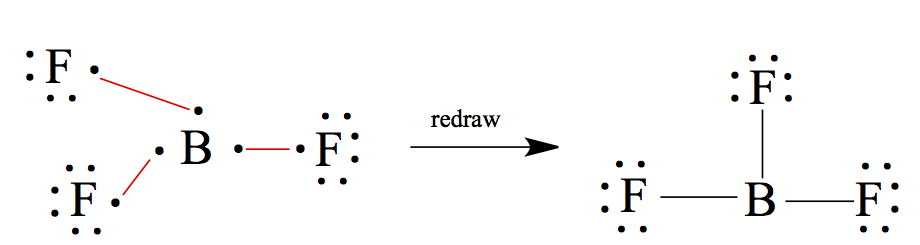
BF3: B is group 13, therefore 3 valence electrons, and F is group 17, thus 7 electrons. B should be the central atom.
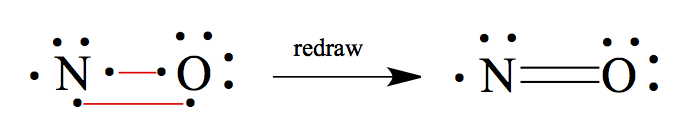
NO: N has 5 valence electrons and O has 6. So, connect two sets of unpaired electrons.
CO: Similar to above, but quite difficult case. Using what we have done, that results in the middle case, where we have a double bond. There are two other structures we can draw, without violating the octet rule.
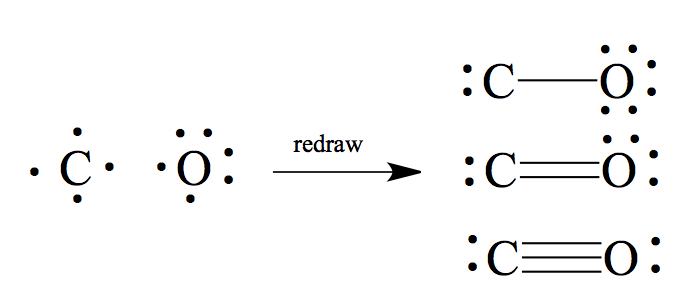
The actual winner is the triple bonded molecule. Generally, C-O single bond is about 1.43Å, and C=O double bond is 1.23Å. Carbon monoxide bond is much shorter, with 1.13Å, and therefore we must conclude that there must be a triple bond to be consistent with the bond length.
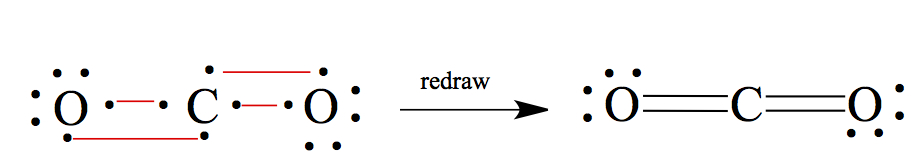
CO2: If you have more than two atoms, less electronegative element becomes the central atom, except for H. In this case, C should be in the center.
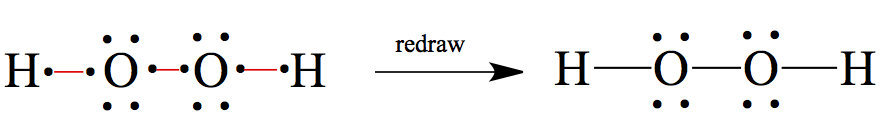
H2O2:
For ions
The following examples show how you might go about constructing ionic structures.
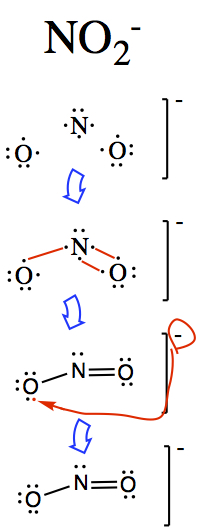 |
|
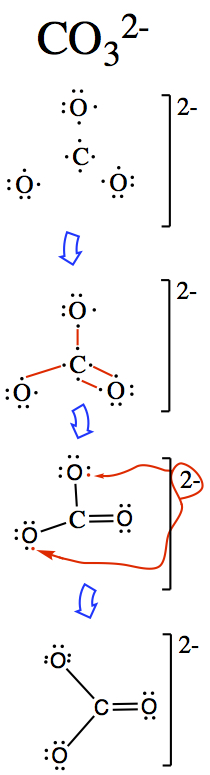 |
|
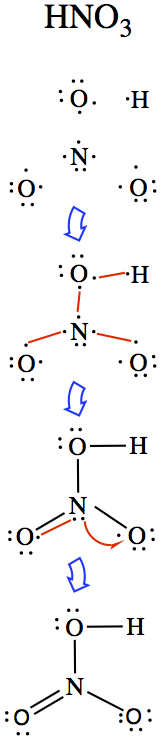 |
Temptation is to do the following: 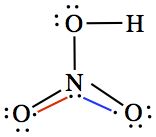 This results in 5 bonded nitrogen which has 10 electrons around it, hence
it is no longer satisfying octet.
This results in 5 bonded nitrogen which has 10 electrons around it, hence
it is no longer satisfying octet.
|
For larger molecules
There are many different structures one can draw for a given formula. For example, C3H4O, I found seven different structures, called structural isomers, as shown below.
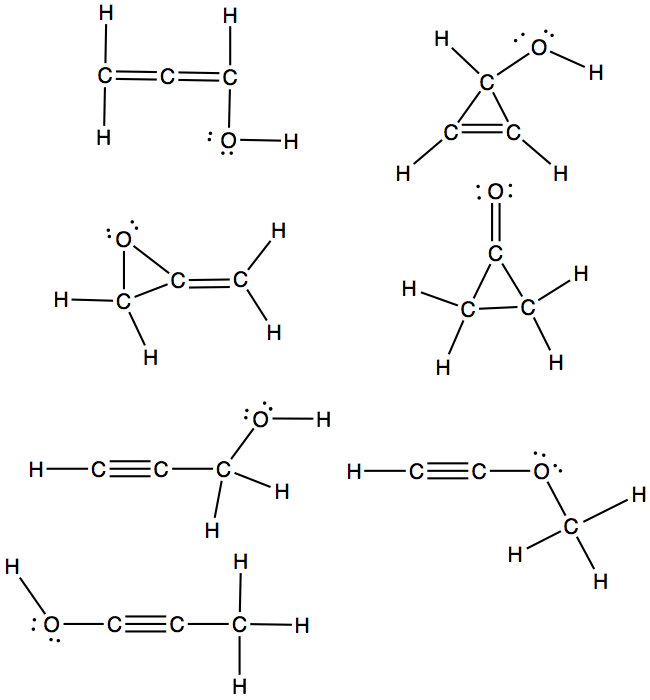
If the formula is C3H6O, then there are 7 (could be more) isomers!! These are all application of connecting dots, and they all satisfy octet!
For molecules containing 3rd row elements and beyond
It is possible for 3rd row elements and heavier elements to have more than 8 electrons around the central atom. We call this expanded octet. For example, the structure of SF4 is shown below.
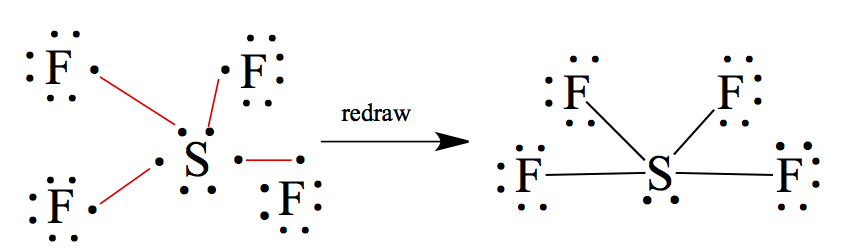
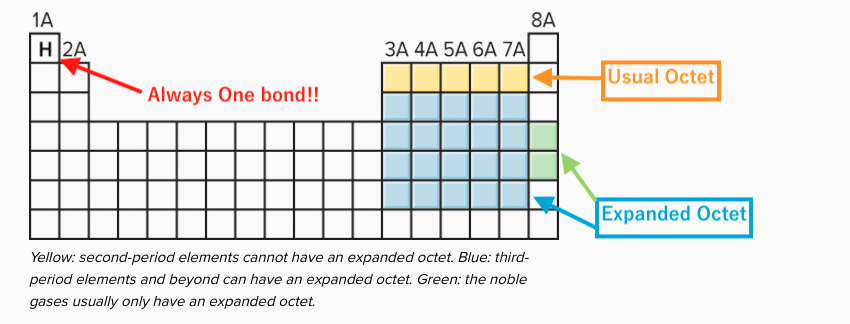
The following diagram will help you determine if the expanded octet should be used in the Lewis structures you might draw.
Resonance Structures
In the case of carbon monoxide structures we examined above, we said that between C and O there should be a triple bond, justifying our structure through experimental bond distance.Just by following the usual way of drawing Lewis structure, you should have arrived at the middle picture with a double bond. The two other structures can be deduced by writing resonance structures from the double bonded structure.
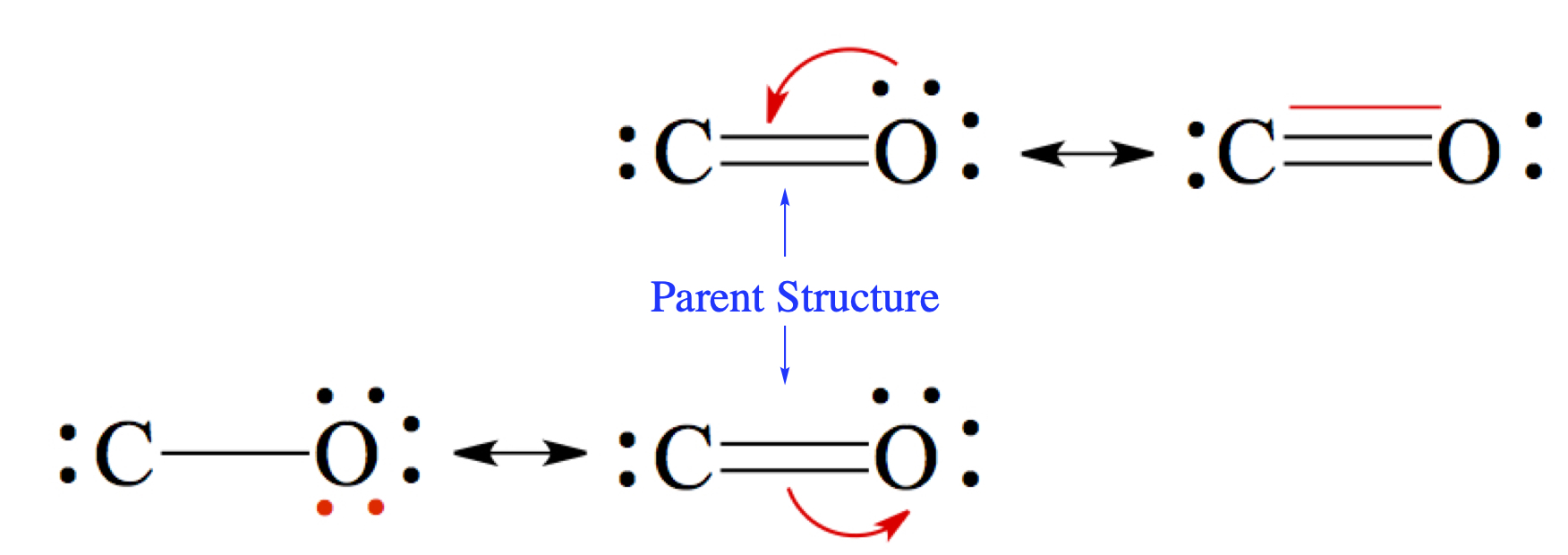
The double-sided arrow, ↔, in chemistry is reserved for resonance structures. The resonance structure with triple bond is constructed by taking one of the lone pair of oxygen and make the pair into a bond between C and O. You notice that there is no violation of the octet rule here. The bottom-left structure is make by moving a pair of electrons from the double bond to the oxygen. Here again, there is no violation of the octet rule. For the C atom, there are only four electrons, this makes C to have + charge and O to have - charge. In the triple-bonded molecule, the C atom has - charge, while O has +. See more in the next section.
1) the resulting structure has the same number of valence electrons
as the parent molecule,
2) Octet rule is followed, except that trivalent
Carbon atom may have the formal charge of -1 and +1, and
3)
the positions of nuclei do not move.
Another example applied to C5H7+ ion.
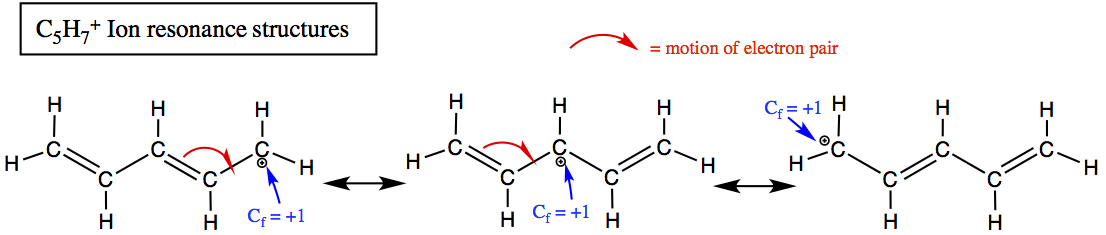
See how the + charge moves within the three resonance structures?
Formal Charges
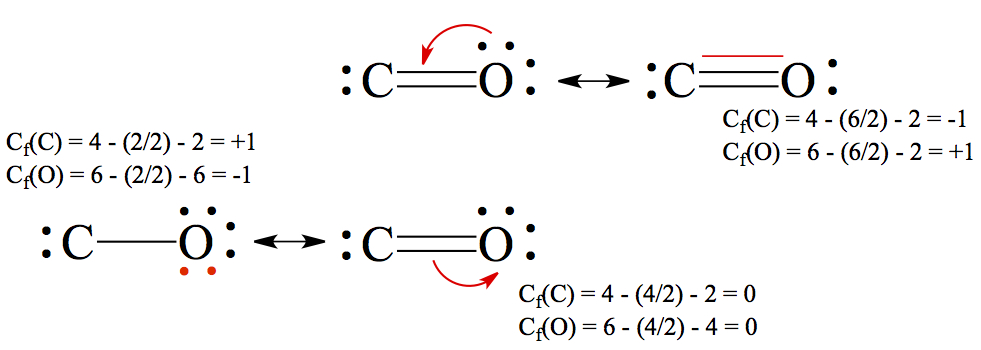
Again, I'd like to bring up the carbon monoxide example. We had single-bonded, double-bonded, and triple-bonded CO, and I made an excuse of having to compare the bond distance to say that the triply bonded CO is perhaps the best resonance structure. Often times, formal charge on the atoms in a molecule is calculated to differentiate the stability of particular isomer.Formal charge, Cf, is a fictitious charge of atoms in a molecule. One can calculate formal charge by using the following equation.
where nve, nbe, and nnb are the number of valence electros, number of bonded electrons, and number of non-bonding electrons, respectively.
Using the carbon monoxide example, we calculate the formal charges of C and O for each of the resonance structure.
These are the structures drawn for the O-C-N configuration.
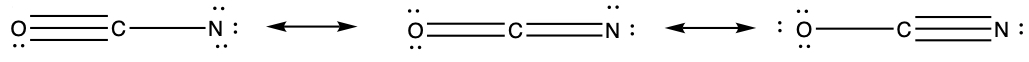
In order to obtain the stability of the ions, one can calculate the formal charges for each atom in the ions. Since all C has four bonds, which means that Cf(C) = 4 - 8/2 -0 = 0. So, calculate Cf for O and N.
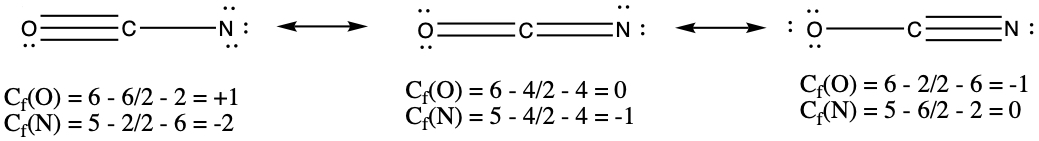
We can assess the stability of molecule by calculating the formal charges of atoms in the molecule, having more than one resonance structure.
This concept is important in Organic Chemistry!
Bond Enthalpy
Bond Enthalpy, BE, is enthalpy change associated with breaking a chemical bond. Thus, BE always has a positive value.Remember that
For example:
One can apply this idea of bond breaking and bond making in approximating the enthalpy change of reaction. After all, reaction is nothing more than bond breaking and bond forming.
Then, the entire reaction can be viewed as a sum of bond breaking (mainly in
reactants) and a sum of bond making (mainly in products).
For example, the enthalpy change of the reaction,
Valence Shell Electron Pair Repulsion Theory
3-D structures of Molecules We now now how to draw a structure of a molecule in 2-D by using connecting unpaired electrons.
Molecules in reality is not two-dimensional. They are actually three-dimensional entity, and have trend to assume certain shapes. In this section we use Valence Shell Electron Pair Repulsion (VSEPR) theory to deduce 3-D structure of molecules. 3-D structure is extremely important, since in many instances, reactivity of molecules can be determined.
From the Coulomb's law, the first equation at the very top of this page, the charges of the same sign have repulsive interaction. Considering the fact that molecules are made of chemical bonds and some have lone pair electrons, there must be bond-bond repulsive interaction or bond-lone pair interaction. The VSEPR theory utilize these repulsive interactions to deduce the 3-D structures of molecules. They key is to minimize the repulsive interactions in a molecule.
VSEPR Theory We must count and classify the number of charge clouds in order to deduce three-dimensional structure.
- # of Bonds: Regardless of having a multiple bond, chemical
bond(s) between two nuclei is considered as one charge cloud.
- # of Lone Pairs: Count the number of lone pair electrons.
- Total # of Charge Cloud: Add the # of bonds and # of
lone pairs.
By using the following table, one can deduce the 3-D structure.
| Charge Clouds | # of Bonds | # of Lone Pairs | Structure | Example | |
|---|---|---|---|---|---|
| 2 | 2 | 0 | Linear | CO2 | 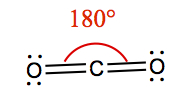 |
| 3 | 3 | 0 | Trigonal Planar | COH2 | 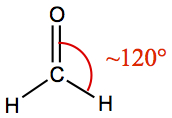
|
| 2 | 1 | Bent | SO2 | 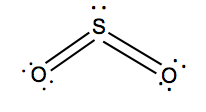 | |
| 4 | 4 | 0 | Tetrahedron | CH4 | 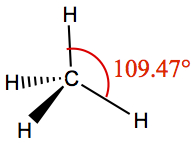
|
| 3 | 1 | Trigonal Pyramid | NH3 | 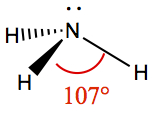 | |
| 2 | 2 | Bent | H2O | 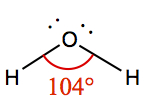 | |
| 5 | 5 | 0 | Trigonal Bipyramidal | PF5 | 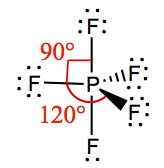
|
| 4 | 1 | See-Saw | SF4 | 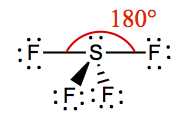 | |
| 3 | 2 | T-shaped | ClF3 | 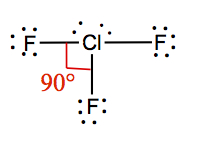 | |
| 2 | 3 | Linear | XeF2 | 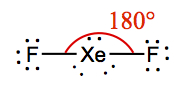 | |
| 6 | 6 | 0 | Octahedral | SF6 | 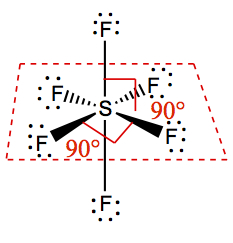
|
| 5 | 1 | Square Pyramidal | ClF5 | 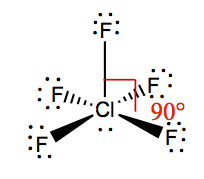 | |
| 2 | 2 | Square Planar | XeF4 | 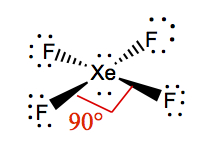 | |
In order for you to determine the 3-D structure, you must first be able to draw a Lewis structure of the molecule so that the number of charge clouds, the number of bonds, and the number of lone pairs are known.
Dipole Moments
We learned above that many bonds are polar bond, determined by electronegativity difference between two atoms. When all bond polarities are added to obtain total polarity, we get dipole moment of molecule.
Remember that Fluorine is has the highest value of electronegativity, χ? Due to electronegativity difference, Δχ, b/w two atoms, bond would polarize.
If the Δχ = 0, then the bond is said to be non-polar.
If the Δχ is non-zero, the bond is polar.
So for molecule N=O, we have
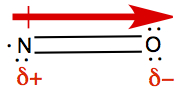 As it is seen, arrow indicates the dipole moment of the bonds between
N and O, and the arrow points toward more negative side, indicated with
δ-, and the other end is more positive side,
δ+. This is so because oxygen is closer to
F in the periodic table than nitrogen.
As it is seen, arrow indicates the dipole moment of the bonds between
N and O, and the arrow points toward more negative side, indicated with
δ-, and the other end is more positive side,
δ+. This is so because oxygen is closer to
F in the periodic table than nitrogen.
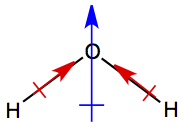 Adding all Polarities of bonds →
molecular dipole moment, μ
Adding all Polarities of bonds →
molecular dipole moment, μ
The bond dipole moments of O-H bonds (red arrow) in water points toward oxygen atom. When these two bond dipoles are added together, molecular dipole moment (blue arrow) is produced. As in the bond dipole, the direction the arrow is pointing has partially negative charge, and the opposite end has partially positive charge.
In order to deduce dipole moments of molecule is to know the 3-D structure of the molecule. Then, draw in the bond dipoles (red arrows). We consider C-H and C-C bonds to be non-polar bonds, therefore we don't draw in the arrow for these bonds. After bond dipoles are drawn in, all bond dipole vectors are added, and the resulting vector is the molecular dipole moment (blue arrow ).
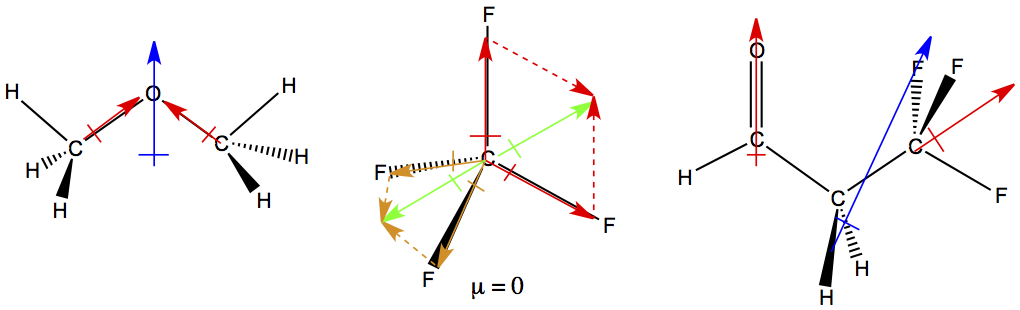
For CF4, two of the red bond dipoles form a resultant green dipole in the plane of the computer monitor. For other two bond dipoles, with orange color, form another green resultant pointing in the opposite direction. And, they cancel each other, therefore the moleuclar dipole moment of CF4 is zero.
Valence Bond Theory and Hybrid Orbitals
When drawing a Lewis structure, we used a pair of valence electrons from each nucleus in the molecule to form a chemical bond.We also talked about formation of chemical bond in H2 from two hydrogen atoms; two 1s orbitals approach each other with overlapping tails, which gave rise to bonding and antibonding orbitals.
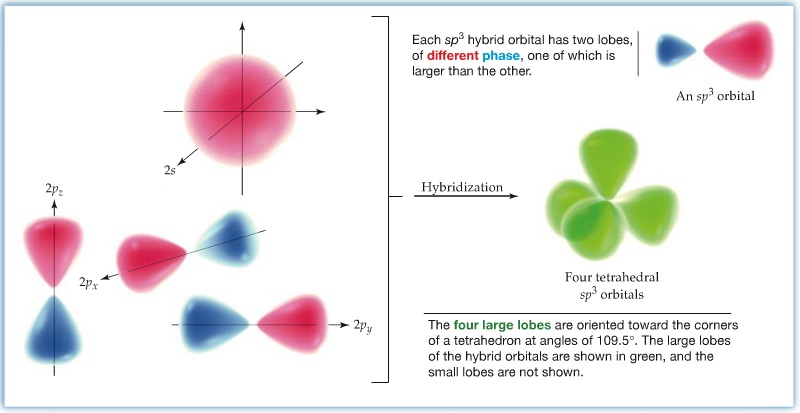
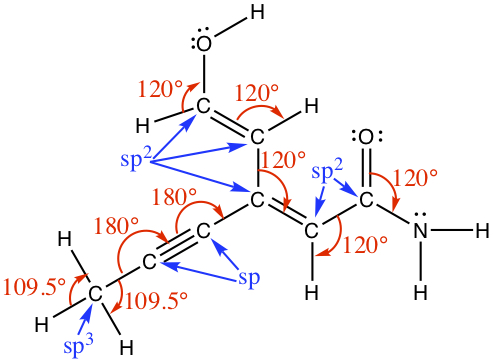
sp3 Hybrid Orbitals: Upon forming a chemical bond, for example in CH4 1s orbitals of four hydrogens overlap with the carbon's 2s and 2p orbitals, because carbon's valency is 4. Since methane assumes tetrahedral structure, each of the four bonds has to be completely equivalent. This means that carbon's atomic orbitals have to be modify to accomodate the four hydrogens' 1s orbitals. The accomodation comes via forming a so-called, hybrid orbitals. Carbon needs to use four of orbitals (2s, 2px, 2py, and 2pz) and the four orbitals mixes to obtain four equivalent atomic orbitals, known as sp3 hybrid orbitals, as shown below. The sp3 orbital overlaps with hydrogen's 1s orbital to form a covalent bond, called σ bond.
The angle between each lobe assumes 109.47° since the sp3 carbon is tetrahedral.
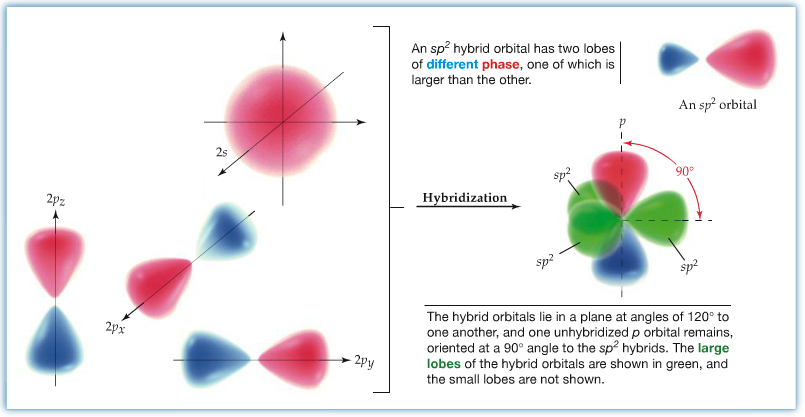
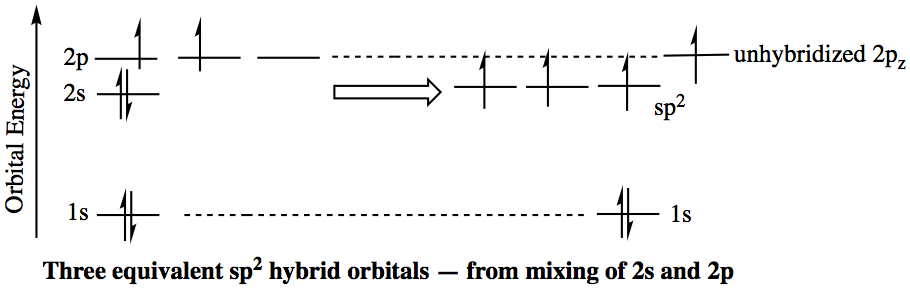
sp2 Hybrid Orbitals: If a carbon atom is in a double-bond environment, such as C2H4 (ethylene), the carbon is hybridized to become sp2 hybrid orbital. As we have seen in the VSEPR theory, double-bonded carbon has a trigonal planar structure with approximate bond angle of 120°. In order to have three σ bond connections, one 2s and two 2p orbitals have to be mixed. This leaves unhybridized 2p orbital. The unhybridized 2p (typically 2pz orbital) orbital is involved as the second bond to another carbon. We call this a π bond . The bond angle between the lobes of hybridized orbitals are about 120°, and each lobe is coplanar. The unhybridized p orbital is 90° to the plane. The sp2 hybrid orbital is shown below.
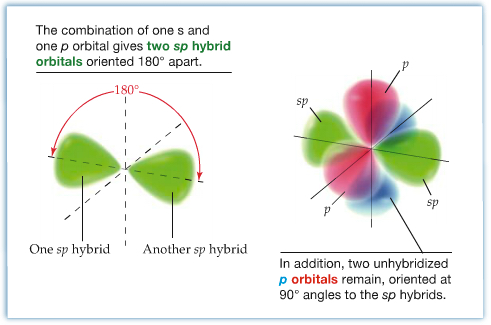
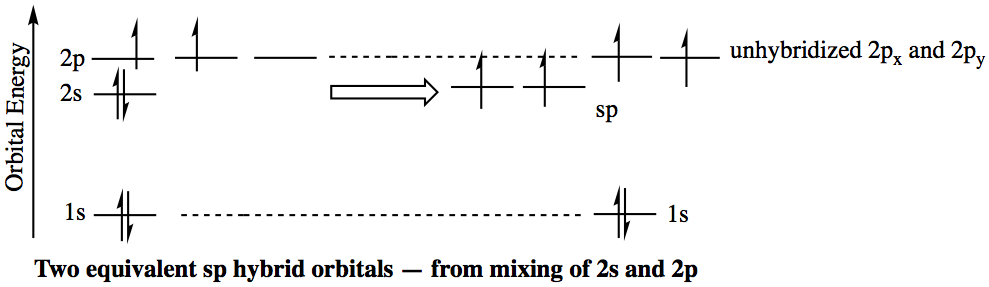
sp Hybrid Orbitals: In this case, a carbon atom is in the triply bonded enviroment. The VSEPR theory predicts that acetylene (C2H2) is a linear molecule (180° bond angle). Each carbon is bonded to two other atoms, hence only two atomic orbitals, namely one 2s and one 2p orbital are needed for hybridization. Two other 2p orbitals are unhybridized. These unhybridized p orbitals are involved in two π bonds.
The following figure shows the bonding between two carbon atoms in ethane (C2H6), ethylene (C2H4) and acetylene (C2H2), for which possess a single bond, double bond, and triple bond, respectively. When a single bond is formed, we refer it to as σ bond. When multiple bond is formed, we have π bond. The π bond is formed by interaction between the two neighboring unhybridized 2pz orbitals. Each 2pz orbital has two lobes with different phase, and for a bonding orbital the two phases match.
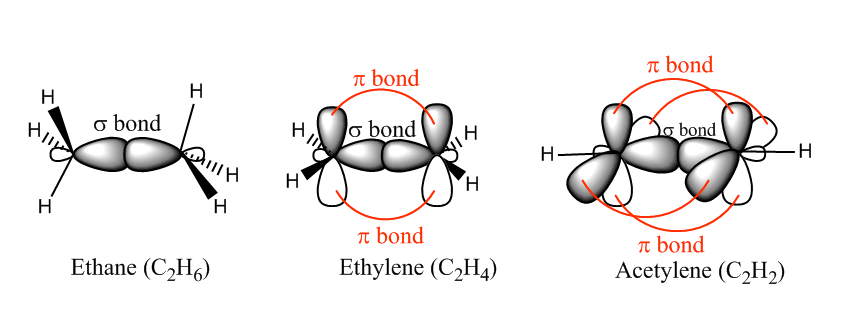
Molecular Orbital Theory
At the beginning of this chapter, we talked about the model of hydrogen moleucle using the Coulomb interactions. Then, from two hydrogen atoms approach each other to have overlapping tails of the two 1s orbitals, which gave rise to a set of molecular orbitals.
Again, for simplicity, we illustrate the formation of molecular orbitals using H2. Each hydrogen atom has one electron in 1s orbital. When the chemical bond forms, the interaction of two waves (1s wave functions) give rise to two molecular orbitals. The bonding orbital, labeled σ is more stable than the original two 1s orbitals. The other molecular orbital is labeled as σ*, which is also known as anti-bonding orbital.
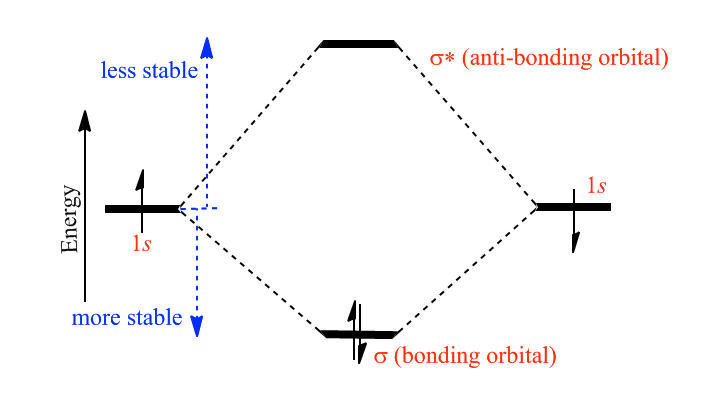
Since the anti-bonding orbital, in the picture below, is higer than the respective atoms, therefore, it is not stable. It means that if electrons are placed in the anti-bonding orbital, the molecule can dissociate. The stability of molecule can be calculated by occupation of orbitals to calculate bond order, OB by using the following equation. Here nbe is the number of bonding electrons and nae is the number of antibonding electrons. If we use this equation for H2-, the bonder order is 1/2, and for He2, OB = 0, and these tell you that H2- molecule is weakly bound and for He2 is expected of no existence.
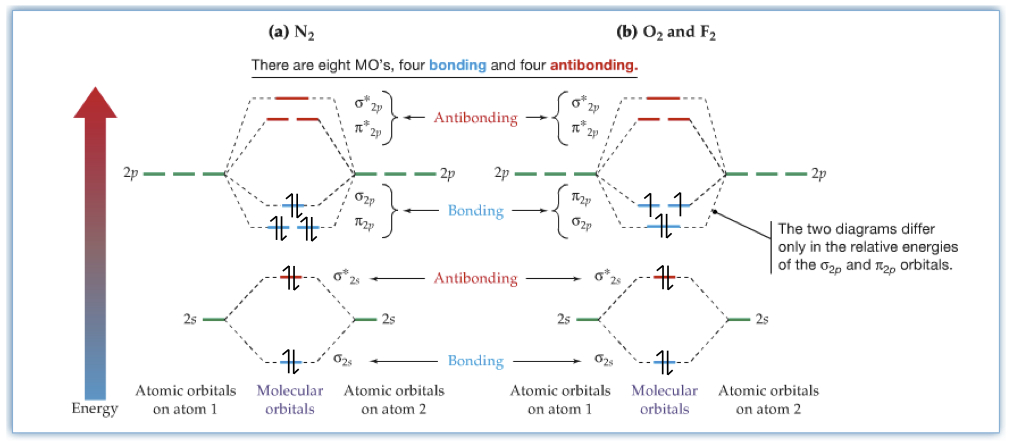
Molecular orbitals of more complicated molecules are very difficult to obtain from a pen and paper. Today with modern computers, we crunch out the molecular orbital calculations. One of the early triumphs of the molecular orbital theory is the diradical character of O2 molecule. When liquid O2 is placed between two magnets, surprisingly the liquid sticks to the magnet. It only means that O2 is magnetic. From Lewis structure, we see that there should be a double bond between the two oxygen atoms, hence there is no magnetic character, called diamagnetic. However, the detailed molecular orbital calculations show that oxygen is paramagnetic, which means that the molecule possesses magnetic character. The following picture compares that the moleuclar orbitals of N2, and O2. The difference between the molecular orbitals is where the bonding orbitals labeled as π2p and σ2p . The order is reversed in O2, which makes the occupation of π2p to be one electron each and they are unpaired.