 |
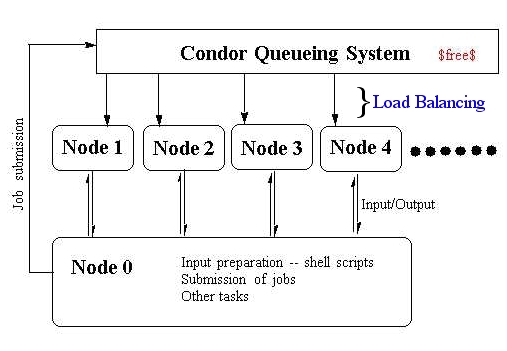
Figure 1. Computing Environment |
Nikita Matsunaga1 and Murali Devi2
1Department of Chemistry and Biochemistry and
2Department of Computer Science
Only 15 years ago, the fastest computer on earth can not compete with what we have on our desktop nowadays. Even with the today's computing power, the computational scientists, including physicists, engineers and chemists, are still pushing the limit of computing by utilizing a better approximation, efficient algorithms, and of course fast computers to obtain more accurate and more realistic models of the nature. In doing so, we often encounter with seemingly impossible prepositions. One example is what is presented here, particularly utilizing minimal effort in establishing the system and virtually no cost if a PC lab is accessible.
If a function is known, then the numerical integration of the function becomes quite trivial. Since the functional form of the equation does not depend on the neighboring points, i.e. one can decouple the integration points to be computed with different computers, say. At the end sum over all results, then you are done. Such system is referred to as an "embarrassingly parallel system".
We have been interested in accurate prediction of transition energies of molecular vibration by utilizing a method called vibrational self-consistent field (VSCF) theory [1], and its mode correlation extension either through perturbation theory (CC-VSCF) [2] and configuration interaction (DPT-VSCF) [3]. The VSCF and related methods have been developed on a quantum chemistry code, called GAMESS [4]. Traditionally in the electronic structure community has been relying on harmonic oscillator model for molecular vibrations, particularly on larger molecular systems. In the VSCF and related methods, the modes must be coupled, and this requires a large number of energies one must compute.
For example, molecular vibration of benzene (C6H6) has 30 degrees of freedom. An implementaion of VSCF in GAMESS requires 16N + 162N(N-1)/2 calculations of a single-point energy, where N is the number of normal modes (here N = 30). It means for a benzene, 111,840 points on the potential energy surface must be evaluated. This translates that if a single-point calculation using a PC takes 10 min., then the computation of the energy points on a single PC takes little over two years! This is of course without any interruption whatsoever! However, if we have six compute nodes, quite modest for such a large problem, running simultaneously, the computaion time is reduced to 4.3 months!! If you can access a lab having 30 computers, which is a realistic number for a PC lab, the amount of time it takes is now reduced to less than a month!! This is the power of "embarrassingly parallel system".
Overall computing environment is shown in Figure 1. It consists of a central machine that does input and other file preparation in addition to the job submission. The central machine here is called Node 0. Other machines are treated as compute nodes (node 1, node 2, ...), and are used only for number crunching purpose.
|
|
Figure 1. Computing Environment |
Each compute node has exactly the same file tree for the GAMESS compuations. In C: disk, we have two directories, "nikita" and "gamess". The former contains the subdirectories, "input", "output", and "batch", and as the names imply for their contents. The "gamess" directory contains executable images of the program GAMESS.
On the node 0, we run cygwin, which is a system of tools that are ports of the popular GNU development tools and utilities for Windows. Many of the useful UNIX commands are implemented for the Windows platform coexisting with the Window applications. We can therefore utilize c-shell script, awk and other favorite UNIX commands and programs simultaneously with the Windows applications such as Word.
The loadbalancing of the compute nodes are done by utilizing a batch queue system called Condor. The Condor queueing system seems to be quite good toward interactive job; the background job is reduced to minimal not to disrupt the interactive job. When a job is submitted to Condor, idle compute nodes are searched, and subsequently the job is submitted to one of the idle compute nodes
On our experimental implementation of the system, we copy all input and batch files to all compute nodes in specific directories. This is due to the fact that the current implementation of Condor does not allow copying files across a network. The files must exist on the local disk. By replicating all input and batch files any job can be submitted to any one of the compute node pool.
It is an easy task to add a node to Condor. It only requires minimal attention for installation, and followed by creation of directories and copying GAMESS program. Therefore, the computing environment itself is also scalable.
Since Condor only runs on NT operating system, it might add to a cost. However, in most academic environment, a PC lab is generally equipped with NT operating system. Of course, the computers must be networked over ethernet. These are the only hardware requirement in constructing the system.
The first step in the computation asks for generation of all input files for GAMESS (See details in CHE606 page or GAMESS on-line manual for input preparation). We use several layers of scripts, C-shell scripts, awk, and others. The input files are copied to a location in each compute node by using the following batch file. More detail is found this way.
| Copying input and batch files across network |
|---|
@echo off echo GAMESS input files are being copied. echo copy C:\nikita\files\input\iso11.inp C:\nikita\input\iso11.inp copy C:\nikita\files\input\iso11.inp \\dell-02\nikita\input\iso11.inp copy C:\nikita\files\input\iso11.inp \\dell-03\nikita\input\iso11.inp copy C:\nikita\files\input\iso11.inp \\dell-04\nikita\input\iso11.inp copy C:\nikita\files\input\iso11.inp \\dell-05\nikita\input\iso11.inp copy C:\nikita\files\input\iso11.inp \\23mzu\nikita\input\iso11.inp copy C:\nikita\files\batch\iso11.bat C:\nikita\batch\iso11.bat copy C:\nikita\files\batch\iso11.bat \\dell-02\nikita\batch\iso11.bat copy C:\nikita\files\batch\iso11.bat \\dell-03\nikita\batch\iso11.bat copy C:\nikita\files\batch\iso11.bat \\dell-04\nikita\batch\iso11.bat copy C:\nikita\files\batch\iso11.bat \\dell-05\nikita\batch\iso11.bat copy C:\nikita\files\batch\iso11.bat \\23mzu\nikita\batch\iso11.bat exit |
The double back-slashes (\\) denotes the networked machine names. As explained above, the input and batch files are needed on each compute node since the current version (6.20) of Condor does not allow manipulation of data over network; the files must reside on a local disk. Furthermore, we can not tell which compute node Condor submits a job next, we must replicate all input files on the local disk.
Job submission is done through the following file_name.sub file. This file specifies the location of the batch file for the job and setting up logfiles (for Condor, not for GAMESS job).
| A script required by Condor for job submission |
|---|
universe = vanilla environment = path=C:\Winnt\system32 executable = C:\nikita\batch\iso11.bat output = iso11.cndr.out error = iso11.cndr.err log = iso11.log queue |
The following file is the batch file to GAMESS run, setting up files needed by GAMESS and executing the program. The environment variables, AOINTS, DICTNRY, PUNCH, and INPUT are set to the name specific to the calculation. Subsequently, the batch file executes GAMESS and reroute its output to specific location. More detail (again!) on how to prepare all these files, click here.
| GAMESS run batch file |
|---|
@echo off echo GAMESS job is being submitted echo set AOINTS=C:\nikita\output\iso11.aoints set DICTNRY=C:\nikita\output\iso11.dictnry set PUNCH=C:\nikita\output\iso11.pnc set INPUT=C:\nikita\input\iso11.inp C:\gamess\gamess.exe > C:\nikita\output\iso11.log echo That's all folks |
These are the only necessary files for constructing an environment for embarrassingly parallel system. It can be foreseen to adapt a web-based system to carry out all of these tasks through CGI scripts. This will be our future work. The following table shows components needed and cost associated.
| Components list and Cost | ||
|---|---|---|
| Components | Qty | Cost |
| PCs with 127 Mb RAM 27 Gb disk running NT | 20 | 0 if a lab exists |
| Condor queueing system | 1 | Free |
| GAMESS Quantum Chemistry Code | 1 | Free |
| Cygwin UNIX tools | 1 | Free |
This is it! One can conceive a system utilizing much broader un-utilized resources on campus, instead of confining the system to one computer lab. In principle, it should be able to scale up to entire campus-wide computing resource without much of inherent problem. Such system is already available commercially at Entropia.com. Their approach is to compile resources all over the world to make one massive parallel computing envrionment by soliciting donation of CPU cycles from organization and from individuals. Their resource is gethered over 34,000 machines world-wide to date and utiltized close to 6,000,000 CPU hours for computing with its peak power delivering 15,948 billion calculations per second. Wow!
Happy computing!
[1] J. M. Bowman, J. Chem. Phys., 68, (1978) 608.
[2] G. M. Chaban, J.-O. Jung and R. B. Gerber, J. Chem. Phys., 111
(1999) 1823.
[3] N. Matsunaga, G. M. Chaban, R. B. Gerber, J. Chem. Phys., in
press.
[4] M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon,
J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nugyen, S. Su, T. L. Windus,
M. Dupuis, and J. A. Montgomery, J. Comput. Chem., 14,
(1993) 1347.
We thank Dr. Steve Elbert of Entropia.com for his suggestion of the problem during a drive in Newport Beach.
Nikita Matsunaga 6/29/02