
Chemistry through computing
Research interest
Anharmonic molecular vibrations
You might have learned in pchem about harmonic oscillator problem. Your professor must have told you that the problem at hand is a model of molecular vibration. Indeed, this is the case. However, the system described by the harmonic oscillator model is highly idealized. In our laboratory, we utilize vibrational self-consistent field (VSCF) method to model molecular vibration more realistic way so that comparison to experiment is facilitated.


molecular diode?
Molecular electornics
According to Moore’s law, the speed of your computer doubles every 18 months or so. If this is to be kept up, people at Intel and others have to shave off the size of a single transistor to yet smaller dimension. It means eventually that the size of transistor reaches molecular dimensions.
anharmonicity
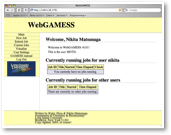
GAMESS is a package of Fortran program that computes properties of molecules by ab initio quantum chemical methods It is freely distributed, and it is a wonderful program to modify for your own research. However, if I want to use GAMESS for my class, such as advanced physical chemistry, students as well as I have a lot of problems. This is because students have to learn unix operating system, and construct a difficult input file. Then, you must be competent enough in computers to submit jobs and retrieve relevant data out of an output that consists practically all numbers!
This is the reason we developed WebGAMESS. This web-based GAMESS management system has following capabilibilities:
° individualized login
° input construction
° check input consistency
° job submissioin
° checks current jobs
° data basing jobs
° visualization of data
° review input and logs
All of our codes are based on freely available script/languages.
The basic knowledge of quantum chemistry is needed to operate WebGAMESS, Only required item is the initial coordinates of atoms in Cartesian. In near future we would like to add a GUI to generate coordinate as well.
Role of surface crossing
In the undergraduate program, you are taught that a chemical reaction is a process that transforms reactants to products via a transition state. For simple reactions, this is a good picture. Life is a lot more complicated than that! How does an excited state decay? How does the spin of electron get flipped during reaction to get diradicals? These are the questions we can not answer from the conventional single potential energy surface picture.
Photochemistry has full of these examples. One of such reactions we are interested in is called spin-forbidden reaction. The reaction starts out having all electrons paired (singlet state). During the reaction, one of the electrons flips its spin to become a diradical state (triplet state). We need to consider both the singlet and triplet potential energy surfaces. The most important feature common to both potentials is the lowest energy crossing point between the two states.
By investigating the minimum energy crossing point, we can learn about what geometry of the molecule the spin is likely to flip as well as probability of the event can be calculated.
Currently, we are examining the vibrational levels of diatomic fluorine molecule in collaboration with Prof. Klaus Ruedenberg and his group at Iowa State University. You may say, “why diatomics now?” The potential energy curve of diatomic molecules to a spectroscopic accuracy are still rather difficult to obtain, particularly the high energy region. The potential energy curve is obtained by the even-tempered Gaussian expansion fitted from the purely ab initio results in which the high-order excitations are extrapolated in the CI expansion. The root-mean-square deviations from the experimental vibrational energy levels are within 10 cm-1.




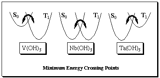

S-T crossing
F2 v=22