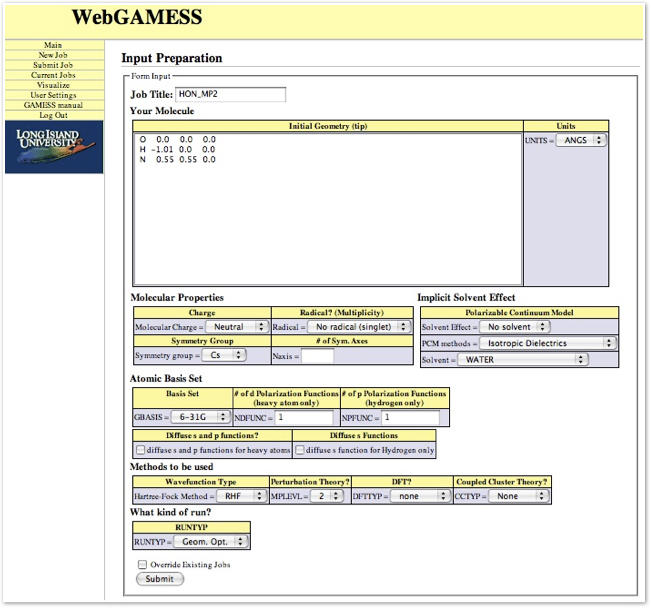
Input preparation
Input is made by point-and-click. Since the choice of keywords are selected from the menus on the browser, typing mistake for the most part is eliminated. Therefore, one can concentrate on the chemistry aspect of the calculations, such as geometry of molecules, which method and basis are appropriate for the molecule, etc. Shown below is an example of the page to construct an input file for a reactive intermediate HON in its singlet state. The calculation is for geometry optimization using the
MP2/6-31G(d,p) method.
First line is a Job Title, which is used as an identifier for individual job. the second line is to
incorporate the atomic symbol and cartesian coordinates. The coordinates of atoms can be difficult to generate at first sight. As a pedagogy, we first calculate atomic positions of simple molecules using trigonometric functions with symmetry considerations. As the molecules become larger, we use commercially available software (Chem3D in our case) to generate initial geometry by optimizing the geometry either with MM2 or semiempirical quantum
chemistry calculations that are built into the commercial software. In near future, we will incorporate a model constructing program written in Java to eliminate the use of the commercial package.
After geometry of the molecule is inputed, one chooses the charge, multiplicity, and symmetry of the molecule.
The basis set is chosen from a set of internally stored basis sets in GAMESS. Choice to add higher angular momentum polarization functions and diffuse functions for anion calculations are also among the choices.
Next is to choose methods or type of wave functions. The Hartree-Fock theory, density functional theory, and Moller-Plesset perturbation theory are chosen from the list. A various flavors of the coupled-cluster theory is available for a single point energy.
The last item is the choice of calculation types. The type of computations that are available are: single point energy, geometry optimization, molecular vibration (Hessian), transition state search, and intrinsic reaction coordinates.
This is all one needs to do, in order to create an input file. By pressing a “submit” button, the program will check your input file by running a so-called “check run”. The program will display 3D image of the molecule inputed, and tells if the input was constructed correctly or not.